
Last week I posted a lecture an incredible by Paul Marik on Fluid Management in severe sepsis. The lecture is the equivalent of a bucket of ice water poured over your head. Now let's give you a towel and discuss.
Want to add a journal club to this flipped classroom?
Then read these pieces in Critical Care:
- Bellomo on Norepinephrine and the Kidney
- Rethinking Resus Goals by Dunser
- Response: Rethinking Resus Goals by Marik and Bellomo
- Pharmacodynamic Analysis of a Fluid Challenge (Crit Care Med 2016;44:880)
The Low-Fluid Volume Early Pressor Experiment has Already Been Tried
It was called standard care 15-20 years ago–patients did not do all that well.
Should we Increase DO2?
We know from that shooting for supranormal DO2 is actually harmful. The original goal-directed therapy trials did not pan out and this may be why.
There is definitely a group of Severe Sepsis patients that are receiving inadequate oxygen delivery. I have treated these patients; I have documented ScvO2s in the 50's and 60's on initial check in a EGDT-type algorithm.
Far more commonly, patients are in pure vasodilatory shock (Jones' trial patients). The latter fact doesn't disprove the former. Using the studies demonstrating the deleterious effects of shooting for above normal delivery doesn't say anything about normalizing patients with low delivery.
Marik's Goals
- Achieve adequate perfusion pressure
- Improve microcirculatory flow
- Limit Tissue Edema
All right on point; how do we get there is the question.
Rivers Trial as a Waterfall?
This is not actually what that trial showed. And the way CVP was used was not actually debunked by the 7 mares article (Note: I'm not advocating you use CVP, I'm just pointing this out! We have better ways to accomplish assessing fluid responsiveness so CVP should be sent to the junk bin)
I will make the utterly blasphemous statement that Dr. Rivers and Dr. Marik are actually in near-complete agreement if you followed both of their protocols explicitly in the ED.
MAP and Association to Survival
Not sure what this is proving: both groups (the flow-optimizers and the desert-inducers) believe in shooting for MAP goals. Patients in whom it is impossible to get the MAP up will die more frequently.
But is it Flow or Pressure that Matters?
I must say, I am still in the tissue flow camp
On to the Glycocalyx
Now this is where stuff gets really interesting. Every day, there is increasing research and more publications on the fundamental role of the vascular glycocalyx. But how do we integrate this clinically?
Chris Nickson tweeted this amazing lecture from Rob Wise. It will explain the Glycocalyx in 5 minutes. It lives on Life in the Fast Lane.
Now as to its relation to fluids in sepsis, we are being told hypervolemia is bad. But if the fluids are leaking from the vascular space are we ever seeing hypervolemia in the vasculature or is the problem whole body volume overload–not if we believe BNP/ANP are the root of the problem, they only respond to vasculature fluids. If we are doing a fluid responsiveness strategy, we should not be seeing the vascular overload.
Do balanced vs. unbalanced fluids have a different effect on the glycocalyx in Sepsis? I have no answers to any of these questions.
ANP/BNP damage the Glycocalyx
But are we causing Myocardial Wall Stress to generate these peptides. Well here is where things get interesting. If you really start thinking this through, CVP EGDT style would make more sense–its use solely to measure RA/RV overdistension rather than fluid responsiveness. Not advocating this in any way, just something to think about.
And is a majority of the BNP coming from fluid resuscitation or from sepsis-induced cardiomyopathy? Is the latter why higher BNP levels are associated with worse outcomes?
Do Crystalloids Stick Around?
I'm not sure, but the Bark Study quoted was a study in septic rats. [cite]23318490[/cite]. This study, if it has any practice-changing value, would be to make one consider albumin. Clinically this lacks all face validity. Start doing IVC exams on your patients, fill until there is no resp collapse. Come back in a few hours, they will have stayed fuller than when you started by a significant amount. Some of this fluid is sticking around.
Let's Talk about Marshmallow People and the Inflammatory State
I'm not sure anything we do makes a damn bit of difference when it comes to patients holding on to water if you are using a reasoned fluid responsiveness strategy, but I am open to new data telling me I am wrong.
Excess Fluid Increases Mortality
This is when the lecture really diverges from clean cause/effect progression. Associations are being taken for causation, so let's spend a bit of time on these:
I'm going to ignore the experimental trials and move to the clinical.
- Fluid Balance doesn't equal fluid administration
- Fluid balance associations certainly do not equate to fluid administration causation (SOAP, etc.)
- CVP associations with fluid administration–Is Dr. Marik, the ultimate CVP debunker, actually equating these two?
Optimal survival occured with a 3-liter positive fluid balance at the 12 hour mark?
FEAST Trial
Dr. Marik's statements on this one need to be looked at carefully. The FEAST trial was an amazing accomplishment with results contradictory to what we all expected. Giving severely ill kiddies fluid increased mortality. But what doesn't get discussed at all is this little article:
subgroup analysis (BMC Medicine 2013, 11:68 ) of why pts actually died revealed CV collapse as opposed to the expected volume overload.
Norepi to tense the Tank
- Critical Care 2007, 11(Suppl 2):P37
- Crit Care Med 2011;39(4):689-94
- Critical Care 2010, 14:R142
- Crit Care Med. 2012;40(12):3146-3153
- Crit Care Med. 2013 Jan;41(1):143-50
Then go read this editorial to get some perspective on the issues involved. [cite source='pubmed']23269148[/cite]
Marik-Phillip Curves
Would indicate that fluid accumulation in the lungs occurs only on the flat portion of the Starling Curve
Should we be using Fluid Responsiveness on all Severe Sepsis Patients?
I think we should. I think we should stop blind fluid loading; it will take a while for us to get there in many EDs.
My modification of Marik's Algorithm:
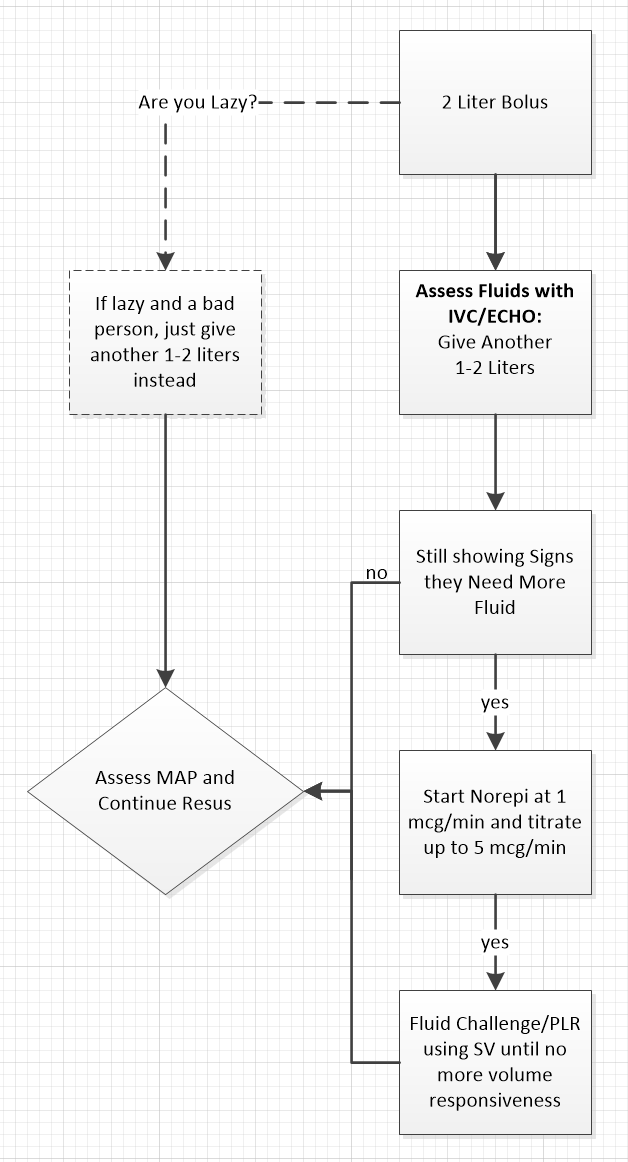
Where are the Endpoints and Is Lactate Clearance is Flawed?
I have already addressed this in a wee when Marik's article was published. See that link if you want all the nitty-gritty details, but here is the essence. Anyone who has talked about lactate clearance for the past decade has already known and perpetrated that the lactate elevation in severe sepsis is not solely from tissue hypoperfusion. We know this. This fact in no way changes that elevated lactates mirror the inflammatory response, and strategies that cause it to clear (probably to near-normal, rather than just 10%) are associated with improvement and decreased mortality.
What about Microcirculatory Endpoints?
Anyone have any good ones? I want one, I want one…
Sepsis Mortality goes Down if you Care!
Paul actually sent me this study. [cite source='pubmed']24201173[/cite] Control arm mortality has dropped consistently since 1991. If you care and do a modicum of resuscitation, I think you are seeing a vast majority of the benefit. Our Sepsis Collab data has shown the same thing.
Read the Comments of the Marik Post
Some amazing stuff buried in the comment section including an inotropy first pathway and the USCOM Device
My Thanks
to Chris Nickson, Cliff Reid, and Minh Le Cong for pre-publication peer review and many helpful comments
Now on to the Podcast…
Additional New Information
More on EMCrit
Additional Resources
You Need an EMCrit Membership to see this content. Login here if you already have one.
Professor
Nassau University Medical Center
No conflicts of interest (coi).
- EMCrit 373 – Mike Weinstock with another Critical Care Bounceback: “Asymptomatic Hypertension” - April 18, 2024
- EMCrit Wee – Ross Prager on 10 Heuristics for the New ICU Attending - April 13, 2024
- EMCrit 372 – FoundStab Intubation SOP - April 5, 2024
I am so eager to listen to the podcast….gonna be my treat later. However one thing I want to throw out that is on topic. Methylene blue. I would love to hear some thoughts on please. Have you guys here ever used methylene blue as a pressor in sepsis?
only tried it a couple of times, will do a wee about it.
Awesome. Would love to read it.
Methylene blue putatively scavenges nitric oxide, which is overproduced by the iNOS isoform (as opposed to eNOS which is inhibited in sepsis). The iNOS form is probably most active in the larger vessels. Thus giving methylene blue should increase your MAP. This may make you feel better but you are not increasing microvascular perfusion and studies tend to bear this out. In contrast, in true vasoplegia like after on-pump CABG, where there is generalized vasodilation but apparently functioning microcirculatory mechanisms, methylene blue will not impair tissue perfusion and works quite nicely. But then one could also use high(er) dose pressors… Read more »
Awesome,
Interesting…but my understanding of the literature is that their are some contradictory findings regarding tissue perfusion and role of enos and inos. Mont studies seem to be in knock out mice and rodent models. I suspect you are correct that it is increasing Bp due to large vessel affects and maybe so what, however it seems less toxic than our true pressors we use. Thanks for the reply…it is really a treat to talk bout this stuff.
There not their
Sorry to get off topic a little – the nitric oxide story has been the stuff dreams are made of – can’t see it, difficult to measure, yet it is seemingly everywhere and does everything. Back in the early 90s there was a lot of hope that this was “the” mediator for sepsis, but even in pre-clinical trials the data was equivocal and non-specific inhibitors led to higher mortality. The thought was that iNOS was ‘bad’ and eNOS was ‘good’ and if somehow we could switch off the ‘bad’ and switch on the ‘good’ things would be ok. Which never… Read more »
Thanks…appreciate your input.
Eric, great to have you on the site! Anesthesia Critical Care, correct?
Scott, This is why FOAMed works! The Hegelian Dialectic at it’s best. I think the most profound portion’s of each of your discussions is the need for measuring our success in volume resuscitation. Dr. Marik expounds on giving fluid ounce by ounce and giving enough but not an ounce more, and you comment on the fact that our management of sepsis pts has resulted in better outcomes, largely through close monitoring and CARE of the patient. We cannot lose sight of that fact. Caring for the patient may be as powerful as what we give and how we give it.… Read more »
thanks again, Mike
Hi Scott
You and Paul both agree a supranormal DO2 is bad.So how do we really know when enough fluid is enough, and when someone has reached a cardiac output that is giving normal levels of oxygen delivery? Is it when someone’s no longer fluid responsive (based off whatever measurement we want to use), or is it possibly well before that? I’m not sure if Paul’s flowsheet targeting a MAP>65 is answering that, and maybe targeting fluid responsiveness isn’t either. Does that come down to us finding better measurements of microcirculation, or can we use Scvo2, or something else?
DO2 can be discovered either through surrogates or directly calculated. If you don’t have a cardiac output monitor, then you need to use surrogates:
Is Hb>7
Is SpO2>90%
Is HR reasonable
Then give a reasonable shot towards optimizing preload
Then look at the heart on echo, is it pumping reasonably?
Do you see a decent pulse ox wave form?
Have the pt’s extremities stayed warm?
So in your view, how important is DO2 in critical care and how do we use it? Intuitively oxygenating the blood and delivering it to the tissues is one of our prime goals. Supranormal is bad, very low is very bad… If minimally invasive CO monitoring catches on,DO2 can be trended like a vital sign in sick ED/ICU pts. Given the limitation that DO2 takes supply but not demand into account, is this perhaps our macro-perfusion resus target?
So we have ways to measure DO2, but what I think we’re missing is a way to measure when we have obtained *adequate* DO2 where further fluid boluses are only going to lead to supranormal DO2 levels (where DO2:VO2 is >>1). What I wonder, and maybe I’m wrong on this (let me know), is whether that ratio is possibly met far before patients stop being fluid responsive.
Yes that is the million dollar ?. When their MAP is low it is an easier call. Once the MAP is good, much tougher. I don’t have that answer.
I think there is no doubt that we can achieve more than adequate levels of DO2 long before we reach the limit of fluid responsiveness provided that somebody has reasonable cardiac function. We are all sitting here reading this nowhere near our limit of fluid responsiveness but with more than adequate DO2. The problem comes in the patient with low inotropy where they show minimal response to fluid challenge, exactly as Starling’s law predicts. In this situation it is quite possible to more than exceed their limit of fluid responsiveness without providing an adequate DO2. In most cases of adult… Read more »
Scott, great podcast. I am not a physician but a pre-hospital provider (Paramedic). I was very interested in the part of the podcast discussing Norepinephrine and its use is Septic shock. When I was going through my training in the mid 1980’s this vasopresser was thought poorly of for use in any kind of shock. It is now being widely used in most hospitals. What has changed?
Jeff, the levophed leaves them dead paradigm was based on the fact that norepi was only given as a hail mary when the pt’s fate had been sealed. The evidence has lead to many of us to stop using dopamine.
Any future for Isolyte as a resuscitative fluid in sepsis? pH 7.4 and electrolytes balanced to more normal physiologic levels seems appealing when compared to NS.
It is just like the plasmalyte and normosol I love. Just a bit more expensive though than NS or LR.
Wow the podcast was awesome, thanks. Few things…. Please correct me if my logic seems off or makes no biological sense.. ! We have spent a good amount of space talking about fluid administration, monitors to estimate adequate hemodynamics, and mortality markers. My brain seems to tell me the following. I think this way and this is how I see the whole thing. Being an organic chemist prior to medical school, I am used to the physical inorganic chemist (Jedi vs darth’s … Had to throw in the Star Wars humor) folks giving me a hard time because us organic… Read more »
great comment my friend. Yes, at the end of the day your clinical judgement is prob. just as good as all of this other crap. One of the pathologies of severe sepsis is they lose the ability to mobilize back into the vasculature a bunch of the fluid lost into the interstitium. At least that was always my understanding, Paul will probably tell me I’m wrong.
Back in the 70s there was a lot of interest in mechanisms of fluid transport in the body and there was the idea that the ‘shocked’ patient (depending on etiology) was fluid down – including the cellular compartment, which can swell and shrink. The idea, conceptually, which I still adhere to, was that one would try to resuscitate the cellular component of the body with crystalloid first (and yes I use NS. Or LR. or whatever’s closest) and once replete the idea was to shift to a colloid to avoid flooding the extravascular (or 3d space) space because it made… Read more »
I’ve often wondered why ICU docs don’t pursue this aspect more vigorously. We are getting good at aggressively resuscitating, but a lot of units are happy to just potter along in the days and weeks post sepsis without pursuing aggressive DE-resuscitation. Why not? Aiming for deresusciation endpoints is at least as key as the “sexy” bit at the start with all the lines, tubes and pressers. We’ve been doing this for years in our unit, and our length of stay, mortality and ARDS incidence outcomes are great as a result. Aiming for a negative balance at day 3, tight electrolyte… Read more »
John,
I have seen the same and you are absolutely right. I love the way you phrase it above.
You are correct, Scott 🙂 I agree with the above gentleman. As our immediate mortality from sepsis has declined (due to EGDT? Surviving Sepsis Bundles? Recognition and early antibiotics? (probably)), long term outcomes haven’t appreciably changed. I don’t have the figures but I think most of our septic deaths now are at hospital day 10-30. As far as I can tell, we have no theory about what to do with patients here hemodynamically or otherwise. Yeah, we try to get them fluid negative, have flirted with beta blockers, steroids, etc, and we put all sorts of monitors on them, but… Read more »
In the UK we have about 1/7th of the ICU capacity of the use per-capita. As a result, septic patients on wards often won’t get near ICU until they received 5+L, and probably abnormal saline to boot. In the UK these are probably the high risk patients that need vassopressor far earlier, but without the resources to provide it early enough. This could be mitigated if we were able to assess fluid responsiveness out side ICU with systems such as uSCom or NICOM. We don’t keep patients in ED for this sort of care (4 hour max stay in ED… Read more »
that sounds rough. Though as I mention at the end, 3 liters, abx, and+- pressors is probably >95% OF THE BATTLE.
We have the same 4 hour limit in ED in Australia – in theory anyway. But that’s plenty of time. 80% of our septic patients in ED are haemodynamically optimised in 1 hour and 95% within 90 minutes. By the time they reach ICU a couple of hours after admission I will usually have gone home! Maybe that is the real answer to why our SS mortality is so low at 6% – we don’t let them wallow in hypotensive hell for hours…
So as the case with most things middle ground seems to be the way to go. Striving to achieve physiologic levels as opposed to supra/infra physiologic levels in resuscitation.
Scott would you still resus according to your ACEP talk, given cited literature? Or would you adjust this scheme in anyway?
Fix the Sat
Dial the Rate
Fill the Tank
Squeeze the Pipes
Flog the Pump
Bolster the Crit
Calm the Beast
everything is the same, and I mention the early use of pressors to squeeze the venous side in that talk. The only thing is that bronze medal is prob. ok in the ED, we are probably moving away from empiric fluids after those 1st few hours as being acceptable.
The benefit of using a non invasive CO monitor is it makes things so much easier than flying by the seat of your pants. You titrate noradren to SVR (rather than risking giving too much or too little). You titrate inotropes to inotropy. You accurately assess preload. Etc. it saves you time and worry.
I would no longer suggest just cracking-on in a really sick patient with an airway issue simply because most times it would work out OK. This site has pulled us into a better place than that. There is a better place also with haemodynamics in sepsis.
Agree monitors would be great, unfortunately they currently all have flaws. As to SVR, be wary–it is a calculation, not a measurement and it is predicated on and changes with CO.
Absolutely.
“Cardiac Output” monitors can measure pressure, estimate or infer flow, and attempt to derive SVR. It is, at best, a second level derivation.
Even if we could magically measure it, we’ve no idea what best suits the patient.
This is what you can often seem to radically change the SVR by giving fluid with a CO monitor in situ. The pressure or the inferred CO may change; and the derived SVR will follow regardless of what the “real” SVR may or may not be.
CO monitors are best thought of as a little box of trend measuring voodoo!
-John
yep, if i had the ability to customize these things, I would love to see 1 screen with just SVi and perhaps a small display of SVV (only useful in a small subset of pts). This is assuming the SVi is actually accurate, which I am unsure of with most of the devices out there.
There are a number of points I feel I must pick up on here. Low fluid volume-early pressor therapy was certainly tried in the past, the big problem of course is that its effects were not adequately monitored. Simply using a vasopressor does not cut the mustard in septic shock, because we have to deal with the inotropy issues as well. What tended to happen was that patients would be drastically over-squeezed resulting in a very high afterload in a patient with low inotropy. The left ventricle would just dilate and fail. “Levophed – leave ‘em for dead!” was a… Read more »
Us country docs gotta stick together Brendan. You send the tables, I’ll sort out the beers.
Kinda wishing Jamie had brought an USCOM back to Kangaroo Island to fiddle with. Maybe demo at smaccGOLD?
Now an offer like that is hard to refuse! The tables are winging their way through the ether to you on that worldwide google interface twitterbook thing.
I would have thought smaccGOLD was a real possibility. I could even work out a few comments for your debate like “real airway docs of a certain age can’t even remember where their checklist is!” (For everybody else, see the smaccGOLD program for day 3 – Friday at their website).
Brendan, I would love to look at them as well, you have my email. As to the comment, you very well turn out to be right about your strategy. The thing is you need to actually do the study. Your before and after is quite nice and is just the thing to establish that an irb should accept your research proposal–it is not, unfortunately the kind of proof you need to establish this is any better than all the other stuff we do based on physiology and insightful theory. If there is one thing we have learned from the >15,000… Read more »
Scott, I very much agree with you that most of Marik’s arguments do commit the same logical fallacy again and again. He points to end point, eg. positive net fluid balance, and takes for granted a causal relationship with fluid administration which appears unjustified. However, I do think his strongest and most interesting evidence comes from the FEAST trial. My understanding of your rebuttal of this trial is that the patients didn’t die from fluid overload so Marik’s theory of the particular mechanism of iatrogenicity is incorrect. Sure, but does the ‘why’ really matter? The evidence seems to state that… Read more »
in reply to Susana in regard to FEAST trial what that trial tells me is giving rapid saline fluid boluses to septic shocked kids in Low resource setting without ICU capability of assisted ventilation/invasive monitoring/pressor support , is harmful! Surviving Sepsis guidelines never recommended giving fluids alone..you need the whole package! FEAST Confirms this!! as for the prehospital RCT showing no or little fluid resus is better than liberal fluid resus on traumatic hemorrhagic shock , here it is http://www.nimbot.com/Med/Articles/Journal%20Clubs/2007-01/Immediate%20vs%20Delayed%20Fluid%20Resuscitation%20for%20Hypotensive%20Pts%20with%20Penet%20Torso%20Injuries/Article.pdf this study has never been replicated yet has been cited to justify a permissive hypotensive prehospital strategy. It has nothing… Read more »
Thanks for this post Scott. Paul’s talk was great value for stimulating thinking on the topic and shocking people into taking the decision to prescribe fluids seriously however it really needed to be taken apart in a critical fashion like this. The common ground you both seem to share is you give some fluid and take a look and then give more. I sometimes reckon we are overthinking fluid responsiveness assessment. It’s fun to play with leg lifts and carotid flow times but when I have primary responsibility for a crashing patient I assess fluid responsiveness by giving some fluid… Read more »
In the last 5 years or so, we have had a better understanding of capillary fluid dynamics, particularly in conjunction with an appreciation of the glycocalyx. We now know that the glycocalyx normally ‘traps’ about a litre and half of plasma water in it (due to its hydrophilic chemical composition!) and that normally in the capillaries, there is a central moving layer of plasma and a relatively immobile layer closer to the endothelium….the bit that is bound to the glycocalyx. This explains the differences in measured capillary and venous hematocrit values, and also why Crystalloid : Colloid equivalence is 1.3… Read more »
John,
Fascinating and a more erudite description of the situation than any I have seen thus far. Which John are you? And what is your affiliation?
Hey Scott,
I have always been an avid consumer of the fantastic pearls of wisdom scattered throughout your site….You are doing great work !.
I am an intensivist from India, presently retired from hospital practice, and concentrating on teaching post graduate trainees in Emergency medicine and Critical care.
Keep up the good work, mate..
Regards,
John.
John: Brilliant I agree 100%, one limits tissue edema “By small volume crystalloid boluses and early use of alpha1-agonists” and then closely monitoring the patients response. The days of giving a 30mlk/kg bolus are gone; this leads to iatrogenic salt water drowning. We also need to revise the starling concept; fluid does not move back into the post capillary vascular system by osmotic forces; lymph flow is the primary mechanism that fluid returns to the vascular compartment; so giving colloids to suck fluid into the vascular compartment is doomed. So in the end it is the capillary hydrostatic forces together… Read more »
Fantastic synthesis John!
Chris
John,
Absolutely fascinating. I’d vaguely heard about the glycocalyx and after hearing Paul’s talk, started to dig a little, but as Scott says, fascinating and highly sensical description. Thank you for lifting some of the fog, I will pursue this with even greater interest.
I’d love to discuss this live sometime!
Philippe
http://www.thinkingcriticalcare.com
Very nice summary, John in India. Just want to caution that the glycocalyx seems to function well at very low [alb] and we dont know if bottled albumin is as effective as your own. Not enough evidence yet to crack open the 4% at the first sign of sepsis.
Thank you Tom W, and I completely agree…..I would be extremely hesitant to reach for that bottle of Albumin too. I work in a resource poor part of the world where stuff like Albumin can only be found in the deep pockets of the privileged elite, anyways!! Again on the topic of Albumin, it is good that you mentioned 4%, as opposed to the 20% that Marik mentions in his slide set… I believe that if you are going to use it at all, you should stick with iso-oncotic Albumin and not hyper-oncotic because using the latter is not going… Read more »
Right again about 20%. When thinking through RSE&GM (Woodcock & Woodcock BJA 2012) I thought I had fluffed it when I found Margarson & Soni had demonstrated that 20% HAS increases red cell dilution by more than the infused volume, which seems to prove plasma pi can reverse filtration, disproving Michel/ Weinbaum/ Curry/ Levick/ Adamson. Eureka moment when I realised hyperoncotic 20% will suck water back from the glycocalyx and so deflate it before any reabsorption can occur! I told Mike Margarson about this at a conference, he said he agrees with my interpretation. RSE&GM predicts hyperoncotic solutions are very… Read more »
TOM.. That’s surely a EUREKA thought. My only reservation is that Prof Gattinoni used a 20% solution in his ALBIOS study which showed improved outcome in septic shock. 20% solution does increase intravascular volume by 4 times the volume infused; so the question becomes does this fluid come from the endothelial surface layer.. .. would be an interseting study to do.
So it would appear that the only way a 20% solution can increase intravascular volume when given as a bolus is to draw fluid out of the ESL (glycocalyx). However I was suggesting giving the 20% solution as a continuous infusion (not as a bolus)… this is currently what we do in our ICU at a rate of 10mls/hr; this should negate the dehydrating effect. It would appear to be a bad thing to give a 20% solution as a bolus. It also appears that if a 5% albumin solution is given as a slow bolus rather than a rapid… Read more »
I don’t know why the Rivers protocol always gets held up as an example of the successful use of CVP in guiding fluid resuscitation. Both the treatment and control groups in the Rivers study used CVP (as well as MAP and UOP) in their protocols. The difference in protocol between the groups was in the use of ScvO2 as an endpoint. This translated into higher numbers of PRBC transfusions and more inotropic support in the treatment group. I realize there was also more fluids in the EGDT group, but how much of this was PRBCs? I am anxious for the… Read more »
I agree. The question must be asked though why did the patients need so many PRBC transfusions if there is this massive capillary leak syndrome in sepsis? Their [Hb] would be high wouldn’t it? They only became anaemic due to all the fluid that was given, and crystalloids don’t carry oxygen, although they may affect the rheology of the peripheral circulation leading to (hopefully) better tissue perfusion, but only up to a point. After that, the reduction in Hb offsets the increase in cardiac output and there is no net gain, or even a loss. Now Paul Marik always tells… Read more »
Agree and I have made the same arguments of ScvO2. What I have heard in terms of hidden back story is that many patients in the trial got blood outside of study protocol, i.e treating docs felt pt would benefit from blood. This would explain why so many people got it. Old blood does regain its ability to release a few hours after transfusion. Heaton A, Keegan T, Holme S. In vivo regeneration of red cell 2,3-diphosphoglycerate following transfusion of DPG-depleted AS-1, AS-3 and CPDA-1 red cells. Br J Haematol. 1989;71:131–6. That being said, I can count on one hand… Read more »
I think Marik might be making too general of a statement if he says that stored blood doesn’t offload oxygen to the tissues. Sure, it might not offload as well, but to say it doesn’t offload O2 at all is a bit of an oversimplification. Granted, there are other concerns with PRBC transfusions including immunosuppression, and nitric oxide scavenging leading to endothelial dysfunction, etc. Plus, what Hct do you target? 30 is pretty arbitrary. And assuming your SaO2 is good then ScvO2 just tells you what is going on with your brain and arms. I dont entirely agree with the… Read more »
This is great stuff, and an awesome debate. I’m out of my league in this comment thread, but I wanted to bring something up: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3512479/pdf/2036-7902-4-8.pdf This was an interesting article looking at fluid balance based on diastolic function and it’s relationship to mortality. Basically, they found that in patients with preceding diastolic dysfunction, those with a lower fluid balance had a higher mortality. Those with a “volume overload” (as predicted by a pseudonormalization of diastolic function (presumed PCWP >15 mmHg) actually did better. They also got more fluid!!!!!! This is interesting to me because this is the very group we… Read more »
brilliant addition, Mike
That is a beautiful explanation of diastolic dysfunction and it’s progression and relationship to filling pressures. It wasn’t long ago that ECHO reports would conclude “normal LV function; diastolic heart failure cannot be excluded” and now ECHO is offering a truly sophisticated explaination of the pathophysioligy- fantastic! I’m not sure about drawing any conclusions though about diastolic dysfunction’s relationship to mortality in sepsis (except for the conclusion that starting out with a normal heart is better than starting out with a sick one). The statistics used to hypothesise about differences between grades of diastolic dysfunction and mortality have the feel… Read more »
Scott, thanks for the fantastic Marik lecture and your excellent critical response. One comment re your response. You state the FEAST study “needs to be thoroughly debunked” but I don’t believe you have done this nor does the article by the authors you linked to. If anything that article firmly debunks most of the criticisms of the FEAST study by showing findings such as no difference in harm from boluses in kids with and without anaemia/malaria. While I agree your acidosis theory has merit, you failed to mention the primary theory put forward by the authors in the article you… Read more »
Anand, the thing that needs to be debunked is not the FEAST trial, it was Paul’s assertions based on it in the context of the debate. FEAST was amazing as we discussed. But this trial has little applicability to our patient populations. I will clarify that in the shownotes.
Now as to fluid somehow being a sustained sympatholytic, I can’t see a shred of physiology or literature as to why that would be the case.
new data leads to new plausible theories. In the critically unwell the sympatholytic effect need not be “sustained” to be harmful. It is entirely biologically plausible that a large bolus could undermine the stress response. In addition I understand there is evidence of saline’s hyperchloraemia inducing renal issues in selected sub groups of patients, but I’m not aware of any good data showing causal mortality differences or cardiovascular collapse. Do you know of such data?
in addition, I’ve posted my thoughts on the FEAST trial and the re-analysis here
http://www.emergucate.com/the-feast-study/
What about Nitroglycerine infusion in septic shock. I know this has been studied before and it is very backwards in traditional thinking. This would unmask volume status to allow for fluid resuscitation. This would maximize 2 of Marik’s initial goals ( 2- improved microcirculation and 3- limit tissue edema) If we prevent the dysoxia to the mitochondria we can maybe prevent the glycocalyx injury, reperfusion injury, improved flow. Again I am sure I am way over my head. Would love to hear from what others thinks, form this knowledgeable group.
Seems like there are 3 types of resus we are talking about:
Macro-circ=plumbers approach
Glycocalyx
Micro-circ/mitochondria
Some suggestion that nitro and dobutamine may help the latter, not sure if they help the glycocalyx
John’s reply got its own blog post. I moved it here: https://emcrit.org/blogpost/more-from-john-lymphatics/
Hi Scott
Thank you for these great podcasts. I have listened to the ones on fluid responsiveness and fluids in general with great interest. I am currently writing up a review article on liberal vs. conservative vs. titrating fluid strategies for critically ill patients.
What truly caught my ear was the argument that 500cc+vasopressors was standard of care in the ED 20 years ago. Are you aware of any material documenting this?
Thanks
Mikael Vognsen
Emergency Medicine Researcher, Beth Israel Deaconess Medical Center, Boston, MA