![Fainting, by Pietro Longhi, 1744. Public domain. [https://commons.wikimedia.org/wiki/File:Pietro_Longhi_027.jpg]](https://i0.wp.com/toxandhound.com/wp-content/uploads/2019/11/image20.png?resize=750%2C389&ssl=1)
Endozepines and Idiopathic Recurrent Stupor
From Cholesterol Anesthesia to Anticonvulsant Potatoes and Hepatic Encephalopathy
by Steven Curry, M.D.
Banner – University Medical Center Phoenix
University of Arizona College of Medicine – Phoenix
Phoenix, AZ
@SteveCurryMD
UPDATE – Enjoy this post? Check out some additional information in a new update as of November 30, 2020.
The upcoming traditional Thanksgiving holiday meal along with anticipated postprandial lethargy reminded me of a topic that toxicology fellows should be familiar with and that provides an opportunity to explore some uncommonly discussed issues surrounding GABAergic neurotransmission – at least uncommonly discussed in medical toxicology fellowships.
Medical toxicology fellows recognize that γ-aminobutyric acid (GABA) is the main inhibitory neurotransmitter of the CNS. To say, though, that the pharmacology, regulation, and consequences of GABAergic neurotransmission are complicated and incompletely understood would be one of the greatest understatements of the century. There are countless papers on topics under discussion in this post, and we’ll be jumping around quite a bit between GABA receptors, benzodiazepine receptors, neurosteroids, endozepines, flumazenil, and other topics. I must dramatically condense and simplify information to make things fit in a blog post. But along the way, we’ll come across anticonvulsant potatoes, cholesterol anesthesia, idiopathic recurrent stupor, panic attacks, Munchausen syndrome by proxy, hepatic encephalopathy, and more. Let’s start in the middle of it all with a couple of papers published in the early 1990s.
Idiopathic Recurrent Stupor
In two papers published in 1992 and 1994, Tinuper and colleagues from the University of Bologna and other centers described four patients with some interesting findings.1,2

Four men, ranging in age from 41 to 70 years, presented for care because of recurrent episodes of unexplained stupor and coma. Episodes would begin with drowsiness, confusion, slurred speech, and staggering gait before proceeding to pronounced obtundation and sleep lasting from 2 to 72 hours. Upon recovery, the patients would be asymptomatic and neurologically unremarkable as well as amnestic for the episode. Attacks occurred as often as every 3 days in one man, and as infrequently as five episodes in 3 years in another. During stupor/coma, EEG monitoring showed low amplitude beta waves (about 14 Hz), but no seizure activity. Vital signs were generally unremarkable, as was oxygenation. There was no history of toxic exposures or use of prescription/illicit drugs that could explain their illness. There was also no response to naloxone.
Extensive investigations were unrevealing or normal, examples of which are provided in Fig. 2.

Toxicology studies included screens for sedatives, barbiturates, and benzodiazepines. While sleep studies showed obstructive patterns, there was no oxygen desaturation.
Low amplitude beta activity on an EEG can be produced by sedation/coma from sedatives such as barbiturates and benzodiazepines. I assume this is why it was decided to give IV flumazenil, a benzodiazepine receptor antagonist, and observe what might happen. Each time flumazenil was given, the EEGs would quickly normalize to alpha waves, and the men would awaken, but they would fall back into a stupor/coma again after a short period of time until resolution of the stuporous episode.
![Figure 3. EEG. [https://commons.wikimedia.org/wiki/File:Eeg_alpha.svg], [https://commons.wikimedia.org/wiki/File:Eeg_beta.svg]](https://i0.wp.com/toxandhound.com/wp-content/uploads/2019/11/fig-3.png?resize=500%2C340&ssl=1)
Injection of flumazenil between episodes had no visible effect and did not change their normal, baseline EEG tracings.
In the absence of benzodiazepines, the physicians wondered whether there might be endogenous substances that were accumulating and activating benzodiazepine receptors. Indeed, in vitro assays of stuporous patients’ blood and CSF revealed a substance(s) that could displace [3H]flumazenil from rat cerebral cortical slices. This suggested than endogenous benzodiazepine receptor agonists played a dominant role in the pathogenesis of what was termed idiopathic recurrent stupor (IRS). A general term given to presumed endogenous ligands for benzodiazepine receptors was endozepines, and the authors suggested that “endozepine stupor” would also be an appropriate name for this disorder.
The authors were not unfounded in their consideration of endogenous benzodiazepine receptor agonists. They cited published studies referring to a peptide from the brain named diazepam binding inhibitor (DBI) and to the fact that diazepam is normally found in the human brain. Yes, that’s right, diazepam had been identified in the brains of persons who had not taken benzodiazepines. The authors were particularly interested in a poorly characterized non-peptide that had been named endozepine-4, present in brain tissue. When purified, this substance had been found to act as a benzodiazepine receptor agonist. In fact, in their patients, endozepine-4 levels in serum and CSF during stupor/coma exceeded values from control subjects by 40 to 300 times, respectively, and returned to control values in between stuporous episodes.
Wow. Endogenous benzodiazepine receptor ligands, endozepine-4, diazepam binding inhibitor, and brains normally containing diazepam. Medical toxicology fellows don’t usually hear much about these topics, though you can find most of them mentioned in Goldfrank’s text in various chapters. Let’s develop some foundation for all of this. We need to begin with a brief overview of GABA and benzodiazepine receptors. We’ll come back to recurrent stupor in a while.
GABA Receptors
GABA was first identified as an inhibitory neurotransmitter at crustacean neuromuscular junctions. It remained controversial as to whether it served as a transmitter in the mammalian brain until the late 1960s. We now recognize that GABA acts as a neurotransmitter in about a third of all brain neurons. After release, GABA binds to two main types of receptors.
![Figure 4. [https://commons.wikimedia.org/wiki/File:GABAa_receptor.gif]
[https://commons.wikimedia.org/wiki/File:GABAB-receptor2.jpg]](https://i0.wp.com/toxandhound.com/wp-content/uploads/2019/11/fig-4.png?resize=750%2C460&ssl=1)
GABAA receptors, of particular interest to us today, most commonly comprise 5 protein subunits that form a Cl– channel. Most commonly there are 2 α, 2 β, and 1 γ subunit combinations, with various subtypes of α, β, and γ subunits to choose from. Binding by GABA increases the probability of channel opening, allowing Cl– influx, and hyperpolarization of a cell away from threshold (i.e., the membrane potential becomes more negative).
GABAB receptors are dimers of G protein-coupled receptors which are mainly coupled to Gi/o. We will not say much more about GABAB receptors.
The GABAA receptor contains binding/recognition sites for numerous ligands apart from where GABA binds. Binding at these additional sites enhances or impairs Cl– influx and, thus, GABAergic tone. Examples of agents that act to enhance the action of GABA and/or directly open the Cl– channel, themselves, (sometimes depending on dose) include propofol, volatile anesthetics, barbiturates, benzodiazepines, etomidate, ethanol, and various neurosteroids (Fig. 4). Our interests in this post mainly concern benzodiazepines and neurosteroids.
Benzodiazepine Receptors or Binding Sites
Central (GABAA) benzodiazepine receptors
Sternbach and colleagues at Hoffman-La Roche Laboratories were seeking new tranquilizers in the 1950s, and they identified chlordiazepoxide as a candidate while examining the activity of quinazoline oxide.3
![Figure 5. Quinazoline oxide (left) and chlordiazepoxide (right).
Public domain. [https://commons.wikimedia.org/wiki/File:Chlordiazepoxide_structure.svg]
[https://commons.wikimedia.org/wiki/Category:Quinazoline#/media/File:Chinazolin.svg]](https://i0.wp.com/toxandhound.com/wp-content/uploads/2019/11/fig-5.png?resize=500%2C264&ssl=1)
Chlordiazepoxide was introduced in 1960 (Librium®), and diazepam (Valium®) followed in 1963. By the mid to late 1970s, benzodiazepines were the most commonly prescribed medications in the world. But it wasn’t until 1977 that the benzodiazepine receptor was reported as residing on the GABAA Cl– channel through studies examining the site of radiolabeled diazepam binding. At first, it had been suspected that GABA and benzodiazepines had affinities for the same binding site on the Cl– channel, but expression of recombinant GABAA receptors showed benzodiazepines bound to a separate modulatory site. It is now established that benzodiazepine binding increases the affinity of GABA’s binding site for GABA and increases the frequency of Cl– channel opening in response to GABA binding. The benzodiazepine receptor on the GABA ionotropic receptor (GABAA) was termed the central benzodiazepine receptor, reflecting its location in the CNS. It has been recommended that benzodiazepine receptors should be referred to as benzodiazepine binding sites rather than receptors. While I strongly agree, most all of the papers cited in this post use the term benzodiazepine receptor, so that’s what I’ll generally do here.
Benzodiazepines bind at a site between an α and γ subunit (Fig. 4). GABAA receptors containing α1, α2, α3 or α5 subunits are responsive to benzodiazepine binding, while those with α4 or α6 subunits are relatively unaffected by benzodiazepines. Ligands for the benzodiazepine receptors are generally divided into 3 groups:
- positive allosteric modulators enhance GABAergic inhibition.
- negative allosteric modulators act as inverse agonists to attenuate or decrease GABAergic tone.
- antagonists bind to the benzodiazepine receptor to displace other ligands, but have no activity in and of themselves – flumazenil is the prototypic ligand in this category.
Peripheral benzodiazepine receptors: the 18 kDa translocator protein (TSPO)
Using radiolabeled diazepam to identify benzodiazepine binding sites, a second receptor for benzodiazepines was found in the kidney and in organs especially active in steroid synthesis.4 This receptor was initially called a peripheral benzodiazepine receptor, and then later, a mitochondrial benzodiazepine receptor. We now recognize that this receptor also occurs in the brain and resides on a protein in the outer mitochondrial membrane termed the 18 kDa translocator protein (TSPO); this serves as the current name for the peripheral benzodiazepine receptor.

TSPO serves several functions and forms a complex with other mitochondrial proteins, including the voltage-dependent anion channel (VDAC; mitochondrial porin) and the adenine nucleotide transporter.5,6 Of interest to us, TSPO transports cholesterol from cytoplasm into mitochondria, the rate-limiting step for the synthesis of neurosteroids. In the matrix, imported cholesterol is converted by CYP11A1 to pregnenolone, which undergoes conversion to various additional neurosteroids.4 Agonism of the benzodiazepine receptor on TSPO enhances cholesterol transport and neurosteroid biosynthesis. In the brain, neurosteroids are synthesized in glial cells such as astrocytes, as well as in neurons.
Neurosteroids have their own binding site on the GABAA complex in membrane-spanning regions between α and β subunits (Fig. 4), and there is evidence for additional binding sites, as well.7 Agonism of the neurosteroid receptor enhances Cl– influx and inhibitory tone, both by enhancing the efficacy of GABA at its binding site, and of benzodiazepine receptor agonists at their binding site. At least some neurosteroids can directly open the Cl– channel in the absence of GABA binding. Various steroid pharmaceutical agents, some used in animals (e.g., alphaxalone), as well as volatile and intravenous anesthetics (e.g., propofol, etomidate) act as agonists at or near the neurosteroid receptor.
In reality, there are also excitatory neurosteroids that decrease GABAergic tone and enhance glutamatergic tone at NMDA receptors, but we don’t have time and space to discuss them today. Rather we are focusing on inhibitory neurosteroids that enhance GABAergic tone. Let’s create a figure summarizing information as we go along.

Cholesterol, of course, possesses the general polycyclic steroid nucleus. Below is cholesterol compared with THPROG (tetrahydrodeoxycorticosterone), one of several neurosteroids.
![Figure 8. Cholesterol (left) and THPROG (right). Public domain. [https://commons.wikimedia.org/wiki/File:Cholesterol.svg],[https://commons.wikimedia.org/wiki/File:Tetrahydrodeoxycorticosterone.png]](https://i0.wp.com/toxandhound.com/wp-content/uploads/2019/11/fig-8.png?resize=1383%2C494&ssl=1)
Given the similarity in structure, a curious scientist might wonder whether cholesterol would bind to the neurosteroid receptor to enhance inhibitory tone. In 1927, Cashin and Moravek from McGill University described the consequences of giving IV cholesterol to cats.8

They first noted that 6 mL of a 3% cholesterol solution given IV caused death within 30 seconds. Not a good start. But they persevered and found that animals survived smaller doses and, interestingly, experienced anesthesia with insensitivity to pain. Abdominal surgery could easily be performed without any reaction from the animal. Anesthesia was of short duration and usually lasted 20 minutes before the animal appeared normal again. I have illustrated this study, below.
![Figure 10. [http://clipart-library.com/free-cat-images.html],[http://clipart-library.com/clipart/464619.htm]](https://i0.wp.com/toxandhound.com/wp-content/uploads/2019/11/fig-10.png?resize=500%2C249&ssl=1)
While Cashin knew nothing of GABA or neurosteroids, in 1992 Majewska mentioned her observation that cholesterol altered GABA binding to the GABAA receptor.9 Sooksawate and Simmonds have since presented experimental results strongly suggesting cholesterol competes with neurosteroids for binding to their receptors on GABAA Cl– channels.10,11
Endozepines
The discovery of benzodiazepine receptors in the 1970s immediately led to the question as to whether there are endogenous molecules that modulate GABAergic tone through interaction with these receptors, just as endorphins, enkephalins, and dynorphins interact with various types of opiate receptors. The term “endozepine” has been used in various ways over the years, but generally refers to any endogenous substance that binds to benzodiazepine receptors. The focus was initially on ligands that interacted with the benzodiazepine binding site on GABAA receptors, but we will see that some endozepines may also have an affinity for the benzodiazepine receptor on TSPO.
As we interpret the results of various studies, keep in mind that flunitrazepam and flumazenil bind to benzodiazepine receptors on GABAA complexes, but not significantly to TSPO (peripheral benzodiazepine receptors). Medicinal benzodiazepines vary in their affinity for central or peripheral benzodiazepine receptors, but diazepam and chlordiazepoxide bind to both, while clonazepam and lorazepam only bind to central benzodiazepine receptors. 4-Chlorodiazepam (Ro5-4864) and isoquinoline derivatives of carboxamide (e.g., PK11195) are specific for binding to peripheral benzodiazepine receptors.

Scientists had good reasons to suspect endogenous benzodiazepine receptor ligands were active. As examples, flumazenil had been reported to trigger panic attacks in patients with panic disorders, but not in control subjects.12 Flumazenil had also been reported to reverse or improve EEG findings and behavior in animals and humans with hepatic encephalopathy.13,14 In vitro studies also supported flumazenil affecting responses to GABA in the absence of benzodiazepines. These and other observations, then, suggested that endogenous modulators were acting at GABAA benzodiazepine receptors.
Using radiolabeled benzodiazepines, flumazenil and other compounds in competitive binding assays on brain tissue, a relatively large number of endogenous substances were shown to bind to benzodiazepine receptors. For most of these compounds, though, endogenous concentrations and/or receptor affinities were too low to support normal physiologic activity.
In this post, we will briefly discuss endozepine candidates that have attracted much attention and/or for which there is support for possible activity at physiologic concentrations. We will divide these endozepines into two general groups: peptides (proteins) and non-peptides. The table, below, summarizes the candidates we will examine.

Peptide endozepines
In 1978 two groups of investigators reported the presence of a peptide in rat and bovine brain that competed with [3H]diazepam for binding to synaptic membranes from the cerebral cortex.15,16 In 1983 Guidotti isolated and purified a protein that was named diazepam binding inhibitor (DBI).17 Intraventricular (brain) injection of the peptide in rats produced effects that were inhibited by flumazenil, indicating that DBI was acting at GABAA benzodiazepine receptors.
In the late 1980s, it was recognized that DBI was identical to a protein named acyl-CoA-binding protein (ACBP).4,18 In the Fellow Friday column on Captain Bligh and microvesicular steatosis, I discussed the activation of fatty acids through esterification to coenzyme A by acyl-CoA synthetase to form acyl-CoA esters, with the term “acyl” referring to various lengths of fatty acids. Acyl-CoA esters are very bioactive, and cytoplasmic concentrations normally are kept < 10 nM through association with plasma lipids and protein binding. ACBP is the major protein that binds acyl-CoA esters.
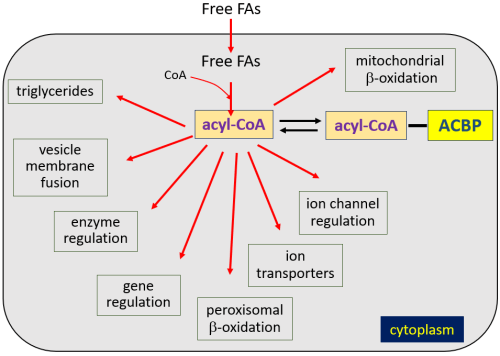
Though ACBP (DBI) is a cytoplasmic protein whose gene does not contain targeting sequences marking it for secretion by recognized mechanisms, studies show that it is, indeed, secreted from cells. While the ACBP (DBI) gene is expressed in brain neurons, it is mainly expressed in astrocytes, and astrocytes are thought responsible for most ACBP secretion. ACBP also undergoes cleavage by an endopeptidase to form additional endozepines: triakotatetraneuropeptide (TTN), octadecaneuropeptide (ODN), and octapeptide (OP) (Fig. 13). Experimentally, ACBP and related peptide secretion can be inhibited by GABAB receptor activation and somatostatin, and stimulated by steroid hormones.4
The human ACBP (DBI) gene shares similarities with those in rats, mice, and other organisms in structure and sequence. Human ACBP, TTN, ODN, or OP have not, of course, been injected into the human brain to observe effects. In vitro studies of ACBP and related peptides have demonstrated both negative and positive allosteric modulation. But overall in vivo behavioral animal studies suggest mainly negative allosteric modulation, with pro-anxiogenic and pro-convulsive activities when injected into the CNS.
Results of behavioral studies, then, suggest that ACBP would be an unlikely candidate for an endozepine that might accumulate to produce recurrent stupor. However, the entire picture is complicated by the fact that ACBP (DBI) also binds to TSPO benzodiazepine receptors to stimulate cholesterol transport and synthesis of neurogenic steroids, which act as positive allosteric modulators. I have added ACBP (DBI) to our figure.

1,4-Benzodiazepines
In 1986, Sangameswaran, De Blas and colleagues described the presence of endogenous nordiazepam (N-desmethyldiazepam) in bovine brains.19 They used an immunoaffinity column with an anti-benzodiazepine antibody in one of many steps of separation and purification and then confirmed nordiazepam’s identity with mass spectrometry. As expected, the nordiazepam competed with [3H]flunitrazepam for binding to rat and bovine brain membranes. Given that their antibody was shown to be binding to nordiazepam and possibly to other benzodiazepines in animal brains, they used immunocytochemistry to examine immunoreactivity in human brains, including an adult human cerebellum that had been stored in paraffin since 1940, long before experimental or commercial synthesis of the first benzodiazepine. The distribution of immunoreactivity was identical in all human specimens to that found in the preserved rat cerebellum. Additional reports have since appeared identifying diazepam and nordiazepam in mammalian brain and blood, including humans. Lorazepam has also been isolated from plasma of drug-free rats.20 That bothers me for some reason.
Documentation of natural benzodiazepines in the human brain suggests endogenous synthesis, dietary sources and/or microbial synthesis. Various benzodiazepines, including diazepam, nordiazepam, and benzodiazepines that have never been commercially produced or marketed are found in milk and a variety of fruits and vegetables, especially in wheat and potatoes.20–24 While plants do not have GABAA Cl– channels, they do possess TSPO, the peripheral benzodiazepine receptor.

In 2007, recognizing that natural benzodiazepines were found in potatoes, Muceniece and colleagues performed an experiment that many of us could only have imagined.25

Muceniece reported that intracisternal or oral administration of a 1:2 dilution of potato juice in normal saline prevented bicuculline-induced seizures in mice. Bicuculline is a GABA antagonist at the GABAA complex used experimentally to produce seizures.
![Figure 17. [http://www.clipartpanda.com/categories/mouse-clip-art-pictures], [http://www.clker.com/clipart-dead-mouse-1.html], [http://clipart-library.com/clipart/464619.htm]](https://i0.wp.com/toxandhound.com/wp-content/uploads/2019/11/fig-17.png?resize=750%2C446&ssl=1)
The authors found a mean diazepam concentration of 38 ug/ kg potato. The fact that potato juice could displace [3H]flunitrazepam binding to brain tissue to a greater degree than that expected from diazepam, alone, was taken to possibly indicate contributions of other potato benzodiazepines in total anticonvulsant effect. Just as interesting, the authors also reported that potato juice diluted 100,000 times inhibited [3H]GABA binding to brain membranes, suggesting that substances that bind to GABA binding sites, themselves, may be present in potatoes; an anticonvulsant effect suggested GABA receptor agonism.
It’s probably premature to jump to conclusions regarding the effectiveness of oral or parenteral potatoes in treating seizure disorders. In general, endogenous benzodiazepines are believed to be at too low concentrations in plasma or brain (or potatoes) to exhibit significant activity in normal conditions. But endogenous benzodiazepines have been reported to be elevated in hepatic encephalopathy. For example, Basile and others found elevated postmortem brain nordiazepam and diazepam concentrations in humans who died from hepatic failure as a consequence of acetaminophen overdose.26 Data from their report are briefly summarized below:

Basile concluded it remained unclear whether nordiazepam, diazepam, and possibly other benzodiazepines contribute to hepatic encephalopathy. He noted that the nordiazepam and diazepam concentrations found in the brains of encephalopathic patients were about the same as after receiving an anxiolytic dose of diazepam. We could discuss many other studies examining endogenous benzodiazepines in the brain and other tissue but must move on to a final group of endozepines before returning to recurrent stupor.
Endozepine-4 and endozepine-2
In 1990, Olmasaa, Rothstein, and others reported the presence of non-protein ligands for GABAA benzodiazepine receptors that did not react with antibodies against benzodiazepines in CSF from patients with hepatic encephalopathy.27 In 1992, Rothstein, Costa, and others better-characterized these ligands using rats and tissue from a human brain.28 Very briefly, HPLC was used to separate substances (endozepines) extracted from brain samples into various fractions that were tested for their abilities to compete with [3H]flunitrazepam for binding to rat cerebellar membranes. Endozepine concentrations were reported in arbitrary units (number of picomoles of diazepam required to inhibit [3H]flunitrazepam binding to the same extent as endozepines in a given fraction). The investigators performed additional assays on these fractions, including endozepine binding affinity to TSPO. Endozepines in a given fraction were also examined for their ability to affect GABA-mediated Cl– currents.
In rat brain extracts, all endozepine activity came off the HPLC column between about 50 and 95 minutes. They found that the 4th peak (fraction), with a retention time of about 75 minutes, had the greatest ability to displace [3H]flunitrazepam. I have roughly sketched their results, below, so you can appreciate the general pattern. The X-axis is the retention time of various fractions that were separated by HPLC, with illustrated peak height being roughly proportional to units of endozepine activity. My sketch consolidates 1 min fractions into single peaks for simplicity. Rat brain data are on the bottom in dark red.

Testing using mass spectrometry excluded the possibility that peak 4 contained diazepam, nordiazepam or oxazepam, though benzodiazepines were found at very low concentrations in some other fractions (< 0.02 ng/g extract), but always too low to affect [3H]flunitrazepam binding. Substances in peak 4 also failed to react with antibodies to benzodiazepines (including diazepam, nordiazepam, and lorazepam) and only weakly bound to TSPO. Some other endozepine fractions, however, did bind more strongly to TSPO. Finally, peak 4 and peak 2 from rats acted as positive allosteric modulators and enhanced GABAergic inward Cl– currents, but did not affect GABA’s binding to its receptor on the GABAA complex. In summary, whatever peak 4 was, it wasn’t a peptide or recognized benzodiazepine, and it appeared to bind to the benzodiazepine receptor on the GABAA complex to act as a positive allosteric modulator.
In order to characterize what one of these endozepines might look like, they needed a lot of brain tissue. A whole lot. So they used 40 Kg (88 lb) of cow brains. Wikipedia says the average cow brain weighs about 0.5 Kg. So, they extracted and purified several micrograms of what chromatographed as peak 2 from brains of about 80 cows.
![Figure 19b. 80 cow brains. Modified from: Vincent van Zeijst \[CC BY-SA 3.0 (https://creativecommons.org/licenses/by-sa/3.0)]](https://i0.wp.com/toxandhound.com/wp-content/uploads/2019/11/fig-19b.png?resize=750%2C400&ssl=1)
They found peak 2 was a single compound with a molecular weight of 265 Da and with suggestions of a quinoline core structure. I have contrasted quinoline with diazepam, below.
![Figure 20. Public domain.[https://en.wikipedia.org/wiki/Diazepam#/media/File:Diazepam_structure.svg], [https://en.wikipedia.org/wiki/Quinoline#/media/File:Quinoline_chemical_structure.svg]](https://i0.wp.com/toxandhound.com/wp-content/uploads/2019/11/fig-20.png?resize=500%2C313&ssl=1)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
What was found in the human brain? Human cortical tissue from a man who died without having consumed a benzodiazepine for at least a year contained similar fractions capable of displacing [3H]flunitrazepam from membranes (upper, green graph, Fig. 19a). However, while humans and rats shared several peaks based on retention times, there was no peak 4 in humans displayed at whatever UV wavelength was used to identify peaks for binding assays. In another portion of the paper, though, the authors described endozepine activity in peak 4 from human brain, presumably indicating there was endozepine activity eventually extracted and concentrated from the fraction collected at about 75 minutes, perhaps using a different wavelength for detection of that fraction. But stop. Why are we even spending all this time discussing endozepine peak 4 with a 75 minute retention time in rat and human brains? Because peak 4 became the famous “endozepine-4” mentioned in reports of idiopathic recurrent stupor, including in one of the two papers we first examined. Let’s return to IRS. But first, we must update the figure we have been creating. In Fig. 21, below, non-peptide, non-benzodiazepine endozepines are shown binding to TSPO, since some fractions of endozepines described by Rothstein did, but their action at the TSPO (peripheral) benzodiazepine receptor remains uncertain (to me).
Back to Idiopathic Recurrent Stupor
Around the time Tinuper and colleagues published the first two papers describing four patients with IRS, they published a third paper in Lancet describing their findings concerning the detection of endozepines in the three men described in their 1994 publication.29

Briefly, serum was collected during stuporous episodes from the three men and collected between episodes from two of the men. CSF (lumbar puncture) was obtained during and between episodes from one patient. Rothstein and colleagues looked for non-protein endozepines, just as they had described in rat and human brain, but, of course, they only had available serum and CSF, not brain tissue. They also performed endozepine assays on serum from seven and on CSF from six control patients. Again, I have sketched a simplified graph summarizing data from their paper (Fig. 23).
First, look what appeared in CSF obtained from one of the men during a stuporous episode. There, coming out of the chromatograph at 75 minutes, was a peak (UV detection at 218 nm), and it displayed a large amount of endozepine activity, meaning it displaced [3H]flunitrazepam from membranes. Based on the retention time, it was identified as endozepine-4.

Keep in mind the original study that characterized endozepine-4 (peak 4) at 75 minutes in rats was centered on brain extracts, not CSF. And in rat (and human?) brain samples, they excluded various benzodiazepines with anti-benzodiazepine antibody assays and analysis with mass spectrometry. Those additional steps did not appear to have been performed on these human serum and CSF samples. And the human brain tissue in their original study, of course, was not from a man with IRS. But there were now control specimens for comparisons. Endozepine-4 was present in low concentrations in CSF from the six controls, grouping around the limit of detection. The mean endozepine-4 concentrations in CSF or serum from a patient during stupor were as much as 300 times higher than control values and returned to control values after recovery. No particular pattern was seen with endozepine-2. It was concluded that excess quantities of endozepine-4 present in the three patients during episodes were high enough to produce stupor, based on diazepam equivalents in the binding assay. The source of endozepine-4 was unknown. Might this be an acquired enzymatic defect preventing degradation of endozepine-4? Or a disorder with increased endozepine-4 synthesis or secretion? Was there a food source high in endozepine-4 content?
IRS recognition grows
Reports of IRS began appearing from other locations in Italy and other countries. By 1998, neurologists from Bologna and others reported 16 new cases and described details for all 20 IRS cases they had been involved with, to date.30 Red stars, below, show the locations of individual patients, indicating no particular commonality to proximity.
![Figure 24. Modified from [https://upload.wikimedia.org/wikipedia/commons/f/f2/Italy_location_map_cropped.svg]](https://i0.wp.com/toxandhound.com/wp-content/uploads/2019/11/fig-24.png?resize=462%2C500&ssl=1)
{kind=link}
These IRS patients all experienced recurrent attacks that varied widely in frequency and had undergone extensive investigations, including immunoassay drug screens for benzodiazepines. And patients displayed the typical EEG findings and responses to flumazenil. Blood or CSF was tested for endozepine-4 levels in nine patients, and all displayed elevated endozepine-4 activity during attacks. One patient displayed endozepine-4 levels in serum > 8,000 times and in CSF > 1,800 times those of control values while symptomatic.
Like all wise scientists, the authors continually questioned their diagnoses and conclusions. They recognized that lorazepam was the only synthetic benzodiazepine that had a similar retention time to endozepine-4 using their extraction/purification and HPLC methodology. Therefore, they specifically looked for lorazepam in serum from four of the 20 patients using a GC-MS method with an ability to detect levels > 5 nmol/ml (1,605 μg/L) but found none. Oral flumazenil at 20 mg/day was tried in five patients, and this was associated with decreased frequency of attacks in the three patients who tolerated it. These investigators had also received reports of IRS patients in Canada and South Africa with elevated endozepine-4 levels. And then things started hopping in Tuscany.
IRS Outbreak
The beautiful, ancient, walled city of Lucca in Tuscany has a long history. Almost 2,100 years ago in B.C. 56, Julius Caesar, Pompey, and Crassus met in Lucca to reaffirm the First Triumvirate.
![Figure 25. Lucca, Tuscany. [https://commons.wikimedia.org/wiki/File:02_Lucca_seen_from_Torre_Guinigi.jpg], [Myrabella] / [Wikimedia Commons] / [CC BY-SA 3.0]](https://i0.wp.com/toxandhound.com/wp-content/uploads/2019/11/fig-25.png?resize=500%2C355&ssl=1)
{kind=link}
The Black Death devastated Lucca and the surrounding area in 1347 and 1348. As plague moved through present-day Italy, about 64% of the population in the countryside around Lucca perished. In about 1998, though, a very different sort of outbreak appeared.
A group of 9 neighbors who lived near Lucca almost simultaneously developed IRS with the well-described EEG findings and transient responses to flumazenil.31–33 Recurrent stuporous episodes lasted 1-2 days and resolved with some transient confusion and amnesia. Patients denied any ingestion of sleeping pills or benzodiazepines, and urine immunoassays were negative for toxins. Dr. Lugaresi and others from the University of Bologna were asked to see the patients and continued to remain suspicious of the possibility of benzodiazepine use in such patients. While endozepine-4 levels were measured using binding assays, more sensitive toxicology screening by HPLC-ESI-MS and GC-MS was available. Thus, samples obtained from patients during stuporous episodes were analyzed, and blood from all 9 patients contained lorazepam . . . from every single patient. An investigation by authorities led to suspects being accused of poisoning their neighbors.34
Of course, lorazepam has the same retention time as endozepine-4, and lorazepam would compete with [3H]flunitrazepam for binding to brain membranes. This strongly implied that most, if not all, of the previously reported cases of IRS represented unrecognized lorazepam toxicity (when elevated endozepine-4 was detected) or effects of other benzodiazepines also missed by urine drug screening that were intentionally or accidentally ingested for various reasons. I note that original attempts at detecting circulating lorazepam by GC-MS in four patients were limited since therapeutic lorazepam levels lie well below the assay’s detection limit of 1,605 ug/L.30
That was pretty much the demise of IRS, though rarely the diagnosis has briefly resurfaced. For example, in 2004 Granot reported on a 71-year-old man with a 16-year history of recurrent stupor and coma who had carried the diagnosis of endozepine stupor for 7 years.35 But after his wife confessed to administering lorazepam to her elderly mother (suspicious nurses), she subsequently confessed to having given oxazepam and lorazepam to her husband over the years, usually mixing tablets into his tea or food. Psychiatric evaluation led to the diagnosis of Munchausen syndrome by proxy. In 2018, Postiglione described using segmental hair analysis to detect triazolam and disprove the diagnosis of IRS.36
Hepatic Encephalopathy
That IRS was actually benzodiazepine toxicity does not necessarily mean that endozepines play no role in normal physiology or disease. This brings us to hepatic encephalopathy. While the pathophysiology of hepatic encephalopathy is certainly complex and not well-understood, there are several lines of evidence indicating increased GABAergic tone may be a significant contributing factor. TSPO (peripheral benzodiazepine receptor) has been reported to be increased in autopsied brains from humans and animals with chronic liver failure, with increased TSPO gene expression seen in animals.14
Neurosteroid concentrations have been found to be increased in the brains of hepatic coma patients (neurosteroids are metabolized by the liver). A general paradigm suggested by some investigators is shown in the figure, below.

With regard to endozepines, we already noted one study in which brain benzodiazepine concentrations were higher in those dying from hepatic encephalopathy than from other causes, and this has been reported in animals, too.26,37 ACBP (DBI) levels are reported to be elevated five times greater in patients with hepatic encephalopathy than in those with liver disease, but with normal mentation.38 But concerning non-peptide, non-benzodiazepine endozepines like endozepine-4, we have no definite answers. Olasmaa reported higher levels in CSF of patients with hepatic encephalopathy than in controls or those with liver disease without encephalopathy.27 In 1998, though, Avallone and others reported that circulating non-peptide, non-benzodiazepine endozepines were inconsistently elevated in cirrhotic patients, whether encephalopathic or not.39 Of course, circulating levels may not reflect concentrations in the brain.
Nevertheless, flumazenil has been reported to attenuate hepatic encephalopathy in animals and humans. A recent systematic review found that flumazenil treatment did not improve survival in patients with hepatic encephalopathy, but there was evidence to suggest it could lessen encephalopathy.40 Others report the effect of flumazenil to be relatively short and not persistent.
![Figure 27. A Thanksgiving meal. [https://commons.wikimedia.org/wiki/File:Our_(Almost_Traditional)_Thanksgiving_Dinner.jpg]](https://i0.wp.com/toxandhound.com/wp-content/uploads/2019/11/fig-27.png?resize=750%2C374&ssl=1)
_Thanksgiving_Dinner.jpg){kind=link}
Conclusion
As we approach Thanksgiving holiday meals, then, I suggest several take-home conclusions:
- GABAergic neurotransmission is incredibly complex. We haven’t even touched on all the other known allosteric effectors or regulators of GABA production and degradation.
- 1,4-Benzodiazepines such as nordiazepam and diazepam normally reside in our brains, and rats scamper about with circulating lorazepam.
- Don’t give IV cholesterol to your cat, except in small amounts.
- Potatoes may be anticonvulsant, but we have yet to figure out the dose.
- Both peptide and non-peptide, non-benzodiazepine endozepines exist, but their roles in normal physiology and disease states remain undetermined.
- Urine benzodiazepine immunoassays commonly miss some benzodiazepines. This should be ancient news to toxicologists.
- Finally, this Thanksgiving most of you will consume potatoes, bread, berries, stuffing and deserts filled with natural benzodiazepines. The turkey and mashed potatoes will be smothered in thick cholesterol-rich gravy. If after the meal you happen to become stuporous, lying on the sofa and difficult to arouse, and someone who recognizes the incredible endozepine and cholesterol load you have experienced awakens you promptly with flumazenil, you can bet that someone slipped you a benzo; maybe a Z-drug.
Postscript
Fellows, here are a few items for discussion and a bit of research.
- Could unrecognized toxicity from zolpidem or related Z-drugs closely resemble recurrent idiopathic stupor? How would toxicity from these agents differ or mimic lorazepam with regard to the following: EEG; response to flumazenil; benzodiazepine immunoassay; benzodiazepine high-sensitivity assay with LC-MS/MS; endozepine-4 levels (peaks) at the 75 minutes retention time? Does zolpidem bind to all benzodiazepine receptors? If you read French, take a look at Huberfeld’s report and see what you think.41
- Why do we miss so many benzodiazepines in screening with urine immunoassays? What role does glucuronidation play with regard to lorazepam? And why do we commonly miss clonazepam and some others? Good questions for board exams.
- The September 5, 2019 issue of the New England Journal of Medicine reports a trial of SAGE-217 in the treatment of major depression.42 Read the introduction of the paper, and if you paid attention today, it should make complete sense to you, except we did not touch much on how neurosteroids affect glutamatergic neurotransmission. What do neurosteroid levels do during depression, and how do they change with therapy? What is the mechanism of action of SAGE-217? Is it a positive or negative allosteric modulator of GABAergic neurotransmission? What do you think an overdose might look like?
Have a great Thanksgiving, and please give us some feedback on Twitter or at this site.
UPDATE – Enjoy this post? Check out some additional information in a new update as of November 30, 2020.
- 1.Tinuper P, Montagna P, Plazzi G, et al. Idiopathic recurring stupor. Neurology. 1994;44(4):621-625. https://www.ncbi.nlm.nih.gov/pubmed/8164814.
- 2.Tinuper P, Montagna P, Cortelli P, et al. Idiopathic recurring stupor: a case with possible involvement of the gamma-aminobutyric acid (GABA)ergic system. Ann Neurol. 1992;31(5):503-506. https://www.ncbi.nlm.nih.gov/pubmed/1317696.
- 3.Bowery N, Smart T. GABA and glycine as neurotransmitters: a brief history. Br J Pharmacol. 2006;147 Suppl 1:S109-19. https://www.ncbi.nlm.nih.gov/pubmed/16402094.
- 4.Tonon M, Vaudry H, Chuquet J, et al. Endozepines and their receptors: Structure, functions and pathophysiological significance. Pharmacol Ther. July 2019. https://www.ncbi.nlm.nih.gov/pubmed/31283949.
- 5.Belelli D, Brown A, Mitchell S, et al. Endogenous neurosteroids influence synaptic GABA<sub>A</sub> receptors during postnatal development. J Neuroendocrinol. 2018;30(2). https://www.ncbi.nlm.nih.gov/pubmed/28905487.
- 6.Bonsack F, Sukumari-Ramesh S. TSPO: An Evolutionarily Conserved Protein with Elusive Functions. Int J Mol Sci. 2018;19(6). https://www.ncbi.nlm.nih.gov/pubmed/29875327.
- 7.Chen Z, Bracamontes J, Budelier M, et al. Multiple functional neurosteroid binding sites on GABAA receptors. PLoS Biol. 2019;17(3):e3000157. https://www.ncbi.nlm.nih.gov/pubmed/30845142.
- 8.Cashin MF, Moravek V. THE PHYSIOLOGICAL ACTION OF CHOLESTEROL. American Journal of Physiology-Legacy Content. October 1927:294-298. doi:10.1152/ajplegacy.1927.82.2.294
- 9.Majewska M. Neurosteroids: endogenous bimodal modulators of the GABAA receptor. Mechanism of action and physiological significance. Prog Neurobiol. 1992;38(4):379-395. https://www.ncbi.nlm.nih.gov/pubmed/1349441.
- 10.Sooksawate T, Simmonds M. Increased membrane cholesterol reduces the potentiation of GABA(A) currents by neurosteroids in dissociated hippocampal neurones. Neuropharmacology. 1998;37(9):1103-1110. https://www.ncbi.nlm.nih.gov/pubmed/9833640.
- 11.Sooksawate T, Simmonds M. Influence of membrane cholesterol on modulation of the GABA(A) receptor by neuroactive steroids and other potentiators. Br J Pharmacol. 2001;134(6):1303-1311. https://www.ncbi.nlm.nih.gov/pubmed/11704651.
- 12.Nutt D, Glue P, Lawson C, Wilson S. Flumazenil provocation of panic attacks. Evidence for altered benzodiazepine receptor sensitivity in panic disorder. Arch Gen Psychiatry. 1990;47(10):917-925. https://www.ncbi.nlm.nih.gov/pubmed/2171449.
- 13.Als-Nielsen B, Gluud L, Gluud C. Benzodiazepine receptor antagonists for hepatic encephalopathy. Cochrane Database Syst Rev. 2004;(2):CD002798. https://www.ncbi.nlm.nih.gov/pubmed/15106178.
- 14.Baraldi M, Avallone R, Corsi L, Venturini I, Baraldi C, Zeneroli M. Natural endogenous ligands for benzodiazepine receptors in hepatic encephalopathy. Metab Brain Dis. 2009;24(1):81-93. https://www.ncbi.nlm.nih.gov/pubmed/19082698.
- 15.Guidotti A, Toffano G, Costa E. An endogenous protein modulates the affinity of GABA and benzodiazepine receptors in rat brain. Nature. 1978;275(5680):553-555. https://www.ncbi.nlm.nih.gov/pubmed/211441.
- 16.Marangos P, Paul S, Greenlaw P, Goodwin F, Skolnick P. Demonstration of an endogenous, competitive inhibitor(s) of [3H] diazepam binding in bovine brain. Life Sci. 1978;22(21):1893-1900. https://www.ncbi.nlm.nih.gov/pubmed/209277.
- 17.Guidotti A, Forchetti C, Corda M, Konkel D, Bennett C, Costa E. Isolation, characterization, and purification to homogeneity of an endogenous polypeptide with agonistic action on benzodiazepine receptors. Proc Natl Acad Sci U S A. 1983;80(11):3531-3535. https://www.ncbi.nlm.nih.gov/pubmed/6304714.
- 18.Farzampour Z, Reimer R, Huguenard J. Endozepines. Adv Pharmacol. 2015;72:147-164. https://www.ncbi.nlm.nih.gov/pubmed/25600369.
- 19.Sangameswaran L, Fales H, Friedrich P, De B. Purification of a benzodiazepine from bovine brain and detection of benzodiazepine-like immunoreactivity in human brain. Proc Natl Acad Sci U S A. 1986;83(23):9236-9240. https://www.ncbi.nlm.nih.gov/pubmed/3024172.
- 20.Wildmann J. Increase of natural benzodiazepines in wheat and potato during germination. Biochem Biophys Res Commun. 1988;157(3):1436-1443. https://www.ncbi.nlm.nih.gov/pubmed/2849941.
- 21.Unseld E, Krishna D, Fischer C, Klotz U. Detection of desmethyldiazepam and diazepam in brain of different species and plants. Biochem Pharmacol. 1989;38(15):2473-2478. https://www.ncbi.nlm.nih.gov/pubmed/2502983.
- 22.Kavvadias D, Abou-Mandour A, Czygan F, et al. Identification of benzodiazepines in Artemisia dracunculus and Solanum tuberosum rationalizing their endogenous formation in plant tissue. Biochem Biophys Res Commun. 2000;269(1):290-295. https://www.ncbi.nlm.nih.gov/pubmed/10694515.
- 23.Wildmann J, Vetter W, Ranalder U, Schmidt K, Maurer R, Möhler H. Occurrence of pharmacologically active benzodiazepines in trace amounts in wheat and potato. Biochem Pharmacol. 1988;37(19):3549-3559. https://www.ncbi.nlm.nih.gov/pubmed/3178869.
- 24.Medina J, Peña C, Piva M, Paladini A, De R. Presence of benzodiazepine-like molecules in mammalian brain and milk. Biochem Biophys Res Commun. 1988;152(2):534-539. https://www.ncbi.nlm.nih.gov/pubmed/3365238.
- 25.Muceniece R, Saleniece K, Krigere L, et al. Potato (Solanum tuberosum) juice exerts an anticonvulsant effect in mice through binding to GABA receptors. Planta Med. 2008;74(5):491-496. https://www.ncbi.nlm.nih.gov/pubmed/18543146.
- 26.Basile A, Hughes R, Harrison P, et al. Elevated brain concentrations of 1,4-benzodiazepines in fulminant hepatic failure. N Engl J Med. 1991;325(7):473-478. https://www.ncbi.nlm.nih.gov/pubmed/1649403.
- 27.Olasmaa M, Rothstein J, Guidotti A, et al. Endogenous benzodiazepine receptor ligands in human and animal hepatic encephalopathy. J Neurochem. 1990;55(6):2015-2023. https://www.ncbi.nlm.nih.gov/pubmed/2172467.
- 28.Rothstein J, Garland W, Puia G, Guidotti A, Weber R, Costa E. Purification and characterization of naturally occurring benzodiazepine receptor ligands in rat and human brain. J Neurochem. 1992;58(6):2102-2115. https://www.ncbi.nlm.nih.gov/pubmed/1315376.
- 29.Rothstein J, Guidotti A, Tinuper P, et al. Endogenous benzodiazepine receptor ligands in idiopathic recurring stupor. Lancet. 1992;340(8826):1002-1004. https://www.ncbi.nlm.nih.gov/pubmed/1357403.
- 30.Lugaresi E, Montagna P, Tinuper P, et al. Endozepine stupor. Recurring stupor linked to endozepine-4 accumulation. Brain. 1998;121 ( Pt 1):127-133. https://www.ncbi.nlm.nih.gov/pubmed/9549493.
- 31.Simini B. Endozepine yarn. The Lancet. February 1998:458. doi:10.1016/s0140-6736(05)78422-1
- 32.Riva R, Lugwesi E, Fanelli R, Mennini T, Chiabrando C. Spinning more yarn: endozepine. Lancet. 1998;351(9109):1138. https://www.ncbi.nlm.nih.gov/pubmed/9660622.
- 33.Lugaresi E, Montagna P, Tinuper P, Plazzi G, Gallassi R. Suspected covert lorazepam administration misdiagnosed as recurrent endozepine stupor. Brain. 1998;121 ( Pt 11):2201. https://www.ncbi.nlm.nih.gov/pubmed/9827778.
- 34.My partner, Frank Lovecchio, has Italian relatives and friends who attempted to gather additional information regarding this event. Persons living in and near Lucca recalled the outbreak, but no record of prosecutions could be easily tracked down. It was explained that new batches of illicit pills would be “tested” by placing them in food and drink of older citizens and observing the effects. Most who recalled the episode suggested this was most likely the case. Anyone with first-hand knowledge is encouraged to comment. In: ; 2019.
- 35.Granot R, Berkovic S, Patterson S, Hopwood M, Drummer O, Mackenzie R. Endozepine stupor: disease or deception? A critical review. Sleep. 2004;27(8):1597-1599. https://www.ncbi.nlm.nih.gov/pubmed/15683150.
- 36.Postiglione E, Antelmi E, Moresco M, et al. Segmental Hair Testing of Triazolam to Unmask a Suspected Case of Idiopathic Recurrent Stupor. J Clin Sleep Med. 2018;14(4):697-699. https://www.ncbi.nlm.nih.gov/pubmed/29609715.
- 37.Basile A, Pannell L, Jaouni T, et al. Brain concentrations of benzodiazepines are elevated in an animal model of hepatic encephalopathy. Proc Natl Acad Sci U S A. 1990;87(14):5263-5267. https://www.ncbi.nlm.nih.gov/pubmed/1973539.
- 38.Rothstein J, McKhann G, Guarneri P, Barbaccia M, Guidotti A, Costa E. Cerebrospinal fluid content of diazepam binding inhibitor in chronic hepatic encephalopathy. Ann Neurol. 1989;26(1):57-62. https://www.ncbi.nlm.nih.gov/pubmed/2549847.
- 39.Avallone R, Zeneroli M, Venturini I, et al. Endogenous benzodiazepine-like compounds and diazepam binding inhibitor in serum of patients with liver cirrhosis with and without overt encephalopathy. Gut. 1998;42(6):861-867. https://www.ncbi.nlm.nih.gov/pubmed/9691927.
- 40.Goh E, Andersen M, Morgan M, Gluud L. Flumazenil versus placebo or no intervention for people with cirrhosis and hepatic encephalopathy. Cochrane Database Syst Rev. 2017;8:CD002798. https://www.ncbi.nlm.nih.gov/pubmed/28796283.
- 41.Huberfeld G, Dupont S, Hazemann P, Adam C, Baulac M, Pierrot-Deseilligny C. [Recurrent idiopathic stupor in a patient: responsibility of exogenous or endogenous benzodiazepines?]. Rev Neurol (Paris). 2002;158(8-9):824-826. https://www.ncbi.nlm.nih.gov/pubmed/12386528.
- 42.Gunduz-Bruce H, Silber C, Kaul I, et al. Trial of SAGE-217 in Patients with Major Depressive Disorder. N Engl J Med. 2019;381(10):903-911. https://www.ncbi.nlm.nih.gov/pubmed/31483961.
Wonderfully complex. I’m very interested in the common features of Quinoline and diazepam, definitely food for thought with regard to Quinoline-induces seizures.
I’m off to eat some potatoes now…..
Very interesting post! To be clear: eating potatoes and other foods high in endozepines does or does not contribute to drowsiness ? 🙂
I’m unaware of any study that has demonstrated sedation from potatoes, to date. But these data are calling for a randomized controlled trial!
I should also mention that if the direct GABAA receptor agonists described in potato juice (apart from benzodiazepines) are proteins, they presumably would be denatured during cooking, Interestingly, though, benzodiazepines are thought to survive cooking.
Thanks for the clarification. Very interesting post I will be sure to have my students and residents read!
Do you have a paper(s)/reference re detection of diazepam in the brains of benzo naive PTS?
“They cited published studies referring to a peptide from the brain named diazepam binding inhibitor (DBI) and to the fact that diazepam is normally found in the human brain. Yes, that’s right, diazepam had been identified in the brains of persons who had not taken benzodiazepines.”
I’d love to read it, looked but cant seem to find anything.
Amazing discussion btw.
Thanks
Hi Braden,
Take a look at references 19, 20 and 26 to begin with.. Thanks for commenting.
Steve
Hi Dr Curry, it’s wonderful to find such a great summary of the “idiopathic recurring stupor” episode. The 1998-1999 public health investigation (and prosecution of the couple who poisoned their neighbors) were extensively covered by Italian media such as La Stampa, La Nazione and especially regional newspaper Il Tirreno, whose archives are online. I can send you the links if you like.
Yes, please post links to those archives! Would love to learn more of the details. Thanks so much. I’ll also send you an email. I would love to add the details to this post.
Thanks.