 Upon first reading the ATHOS-3 trial, I was pleasantly optimistic. Who wouldn't be interested in a shiny new vasopressor? The trial didn't prove much, but it was intriguing. However, it was alarming to hear that the FDA has approved angiotensin II based on it. Precious little evidence is available about this drug. With angiotensin II arriving at hospitals soon, some cautions are in order.
Upon first reading the ATHOS-3 trial, I was pleasantly optimistic. Who wouldn't be interested in a shiny new vasopressor? The trial didn't prove much, but it was intriguing. However, it was alarming to hear that the FDA has approved angiotensin II based on it. Precious little evidence is available about this drug. With angiotensin II arriving at hospitals soon, some cautions are in order.
Caution #5: Angiotensin II has pro-inflammatory activity
Septic shock results from a cytokine storm involving several pro-inflammatory cytokines (perhaps most prominently interleukin-1, tumor necrosis factor, and interleukin-6). Angiotensin II has pro-inflammatory properties, which include increasing interleukin-6 levels:
 Caution #4: Angiotensin II is pro-thrombotic
Caution #4: Angiotensin II is pro-thrombotic

Sepsis usually causes a hypercoagulable state, for example increasing the risk of DVT/PE. At a microvascular level, tiny thromboses may contribute to tissue damage. A pro-thrombotic drug is undesirable in sepsis.
 Angiotensin II increases thrombin formation and impairs thrombolysis (Celi 2010, Dielis 2007). The ATHOS-3 manuscript didn’t report an increase in thrombotic risk when various types of thrombosis were examined separately. However, the FDA package insert reports an increase in the combination of venous plus arterial thrombotic events (13% vs 5%, p=0.02):
Angiotensin II increases thrombin formation and impairs thrombolysis (Celi 2010, Dielis 2007). The ATHOS-3 manuscript didn’t report an increase in thrombotic risk when various types of thrombosis were examined separately. However, the FDA package insert reports an increase in the combination of venous plus arterial thrombotic events (13% vs 5%, p=0.02):
 Caution #3: Blockade of the renin-angiotensin-aldosterone system improves survival in animal models.
Caution #3: Blockade of the renin-angiotensin-aldosterone system improves survival in animal models.

 Based on several mechanisms (including #4-5 above), some investigators have suggested that blocking angiotensin II could be beneficial in sepsis. Animal models confirm that blockade of the renin-angiotensin-aldosterone system may improve survival (Laesser 2004, Hirano 2014)(3). This has led to a bizarre situation where different clinical trials are investigating the efficacy of both angiotensin II and also angiotensin II blockers in sepsis:
Based on several mechanisms (including #4-5 above), some investigators have suggested that blocking angiotensin II could be beneficial in sepsis. Animal models confirm that blockade of the renin-angiotensin-aldosterone system may improve survival (Laesser 2004, Hirano 2014)(3). This has led to a bizarre situation where different clinical trials are investigating the efficacy of both angiotensin II and also angiotensin II blockers in sepsis:
 The fact that opposite hypotheses are being investigated reveals weakness in the basic science behind using angiotensin II for septic shock. If there were definitive pre-clinical evidence supporting the use of angiotensin II, then it would be inconceivable to perform a clinical trial of an angiotensin II antagonist.
The fact that opposite hypotheses are being investigated reveals weakness in the basic science behind using angiotensin II for septic shock. If there were definitive pre-clinical evidence supporting the use of angiotensin II, then it would be inconceivable to perform a clinical trial of an angiotensin II antagonist.
Caution #2: Limited safety data.
FDA approval was based largely on the ATHOS-3 trial, which cannot establish safety:
- This study exposed only 163 patients to angiotensin II, which is underpowered to detect rare adverse events. For example, regardless of your interpretation of the thrombotic risks discussed above, this trial doesn't resolve the issue.
- Adverse events involving the skin were more common in the angiotensin II group (13.5% vs. 7%). This may be a signal of angiotensin II causing digital ischemia.
- Study inclusion required that the patient must have high-output shock based on either a central venous oxygen saturation >70% plus CVP > 8 mm or a cardiac index >2.3. Angiotensin II would be predicted to be less safe among patients not meeting these criteria.
- The dose of angiotensin II was allowed to increase up to 200 ng/kg/min over the first three hours, but subsequently decreased to 0-40 ng/kg/min (figure below). Higher doses over the first three hours were used to prove efficacy, whereas safety was achieved by dropping the infusion dose to a lower level after three hours. This bait-and-switch scheme leaves it unclear whether it is possible to find a dose which is both effective and safe for >3 hours.
 Caution #1: No meaningful efficacy data.
Caution #1: No meaningful efficacy data.

 The primary endpoint in ATHOS-3 was a composite of either achieving a MAP rise of 10 mm or a MAP >75 mm within three hours. Angiotensin II has been known to increase blood pressure for over fifty years. Therefore, choosing a blood pressure endpoint was an easy goal to reach. This composite endpoint seems engineered to guarantee the study a “positive” result (e.g. MAP >75 mm is an artificially high target). When the blood pressure graph is formatted properly, there is no clinically meaningful difference:
The primary endpoint in ATHOS-3 was a composite of either achieving a MAP rise of 10 mm or a MAP >75 mm within three hours. Angiotensin II has been known to increase blood pressure for over fifty years. Therefore, choosing a blood pressure endpoint was an easy goal to reach. This composite endpoint seems engineered to guarantee the study a “positive” result (e.g. MAP >75 mm is an artificially high target). When the blood pressure graph is formatted properly, there is no clinically meaningful difference:
 What is missing from the ATHOS-II trial is improvement in any meaningful patient-centered outcome (e.g. survival, renal function, ICU length of stay). The study protocol did record several additional endpoints including serum creatinine, urine output, time on the ventilator, full SOFA score breakdown by organ, and ICU length of stay (Chawla 2017). Unfortunately these endpoints weren't reported, perhaps because they didn't reflect positively on angiotensin II. It is tantalizing that angiotensin II improved the cardiovascular component of the SOFA score despite making zero difference in the composite SOFA score – arithmetic requires that angiotensin II must have impaired the function of some other organ(s) not reported in the manuscript (1).
What is missing from the ATHOS-II trial is improvement in any meaningful patient-centered outcome (e.g. survival, renal function, ICU length of stay). The study protocol did record several additional endpoints including serum creatinine, urine output, time on the ventilator, full SOFA score breakdown by organ, and ICU length of stay (Chawla 2017). Unfortunately these endpoints weren't reported, perhaps because they didn't reflect positively on angiotensin II. It is tantalizing that angiotensin II improved the cardiovascular component of the SOFA score despite making zero difference in the composite SOFA score – arithmetic requires that angiotensin II must have impaired the function of some other organ(s) not reported in the manuscript (1).
 Vasoconstriction is a tricky business. Some vasoconstriction is needed to establish a reasonable pressure, but excessive or nonselective vasoconstriction can choke off tissue perfusion. The fact that a vasoconstrictor increases blood pressure doesn’t guarantee that it will help patients. For example, 546C88 (a nitric oxide synthesis inhibitor) caused vasoconstriction and shock resolution in a 312-patient MCRCT eerily similar to ATHOS-II (Bakker 2004, abstract below). This seemed like a terrific drug, until it was proven to increase mortality in a larger RCT (Lopez 2004).
Vasoconstriction is a tricky business. Some vasoconstriction is needed to establish a reasonable pressure, but excessive or nonselective vasoconstriction can choke off tissue perfusion. The fact that a vasoconstrictor increases blood pressure doesn’t guarantee that it will help patients. For example, 546C88 (a nitric oxide synthesis inhibitor) caused vasoconstriction and shock resolution in a 312-patient MCRCT eerily similar to ATHOS-II (Bakker 2004, abstract below). This seemed like a terrific drug, until it was proven to increase mortality in a larger RCT (Lopez 2004).

Three comparisons
Comparison of angiotensin II vs. ascorbic acid/thiamine/hydrocortisone
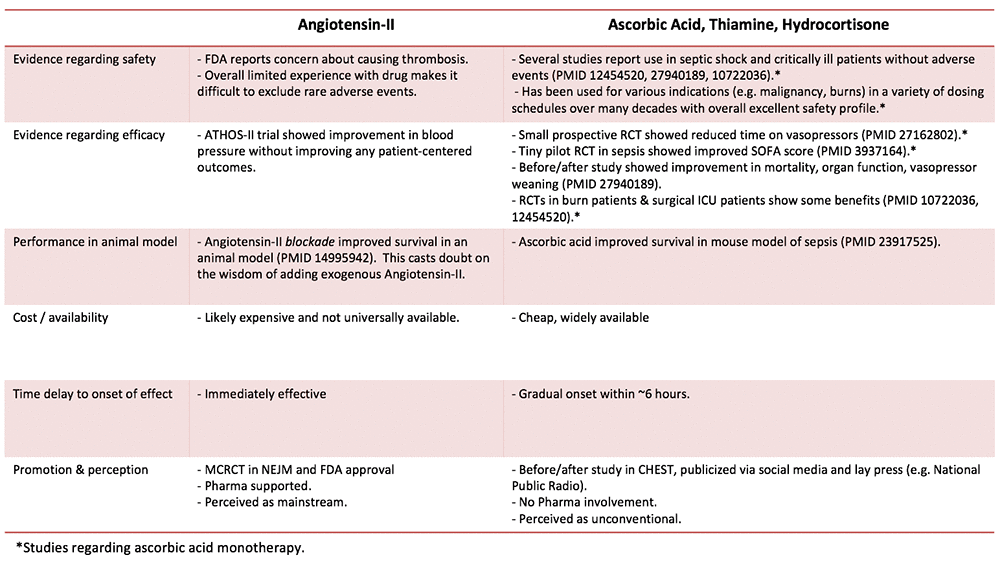
Treatment with ascorbate/thiamine/hydrocortisone is controversial, commonly criticized as lacking evidentiary support. However, there is even less evidence to support angiotensin II. The basic science evidence supporting ascorbic acid is also considerably more robust than the rationale for angiotensin II (more on this here).
Predictions about angiotensin II based on the performance of its next-door neighbor, vasopressin.
Vasopressin and angiotensin II share numerous commonalities:
- Non-catecholamine vasoconstrictors.
- Preferential vasoconstriction of the efferent arteriole of the kidney, potentially increasing glomerular filtration rate and urine output.
- Endogenous neurohormones which may be deficient in septic shock.
- Sometimes effective at low, quasi-physiologic levels.
How useful is vasopressin in septic shock? In the past, I was very enthusiastic about vasopressin based on theory and evidence that it could improve renal function (e.g. prior posts here, here, and here). However, over time this enthusiasm was tempered by growing awareness of problems from excessive vasoconstriction that result from combining vasopressin plus norepinephrine. In some cases, patients developed necrotic toes possibly related to this combination. Currently, my practice patterns have shifted towards reserving vasopressin for patients who are refractory to catecholamine pressors.
That said, there does seem to be a subset of patients who are refractory to norepinephrine and epinephrine, yet respond nicely to vasopressin. These patients often do beautifully with a combination of vasopressin plus epinephrine (“vepinephrine”), with inotropy from epinephrine preventing an excessive drop in the cardiac output:
 Overall, vasopressin is a useful pressor to have in the proverbial toolbox. However, most patients respond nicely to norepinephrine. For such patients, vasopressin is unnecessary and could pose risks of digital ischemia.
Overall, vasopressin is a useful pressor to have in the proverbial toolbox. However, most patients respond nicely to norepinephrine. For such patients, vasopressin is unnecessary and could pose risks of digital ischemia.
Vasopressin's track record should temper our enthusiasm about angiotensin II. In particular, simultaneous use of three vasoconstrictors (norepinephrine, vasopressin, and angiotensin II) could increase risks of digital or intestinal ischemia. However, as with vasopressin, there might be a subgroup of patients who respond unusually well to angiotensin II.
Comparison of angiotensin II vs. phenylephrine
Phenylephrine is often feared, due to concern that it may cause excessive vasoconstriction and drop the cardiac output. This fear isn't really supported by available evidence, which suggests that phenylephrine is rather similar to norepinephrine (2).
 For the many people who avoid phenylephrine due to concerns about dropping cardiac output, it is logical to share the same concern for other pure vasoconstrictors (vasopressin and angiotensin II). It is physiologically illogical to hate phenylephrine while loving angiotensin II.
For the many people who avoid phenylephrine due to concerns about dropping cardiac output, it is logical to share the same concern for other pure vasoconstrictors (vasopressin and angiotensin II). It is physiologically illogical to hate phenylephrine while loving angiotensin II.
 Maybe angiotensin II is a fantastic vasopressor, but currently it's doubtful whether the benefits outweigh the risks. There are several reasons that angiotensin II isn't ready for widespread use:
Maybe angiotensin II is a fantastic vasopressor, but currently it's doubtful whether the benefits outweigh the risks. There are several reasons that angiotensin II isn't ready for widespread use:
- Angiotensin II has pro-inflammatory effects, causing increased levels of interleukin-6.
- Angiotensin II has pro-coagulant effects, which could increase microvascular thrombosis and the risk of DVT/PE.
- Angiotensin II inhibition improved survival in an animal model of sepsis, suggesting that excessive angiotensin II activity can be harmful.
- Angiotensin II hasn't been shown to improve any outcome other than a minor increase in blood pressure. Full stop.
- ATHOS-3 is underpowered to detect uncommon adverse events.
- The rationale for using angiotensin II is very similar to the rationale for using vasopressin. Although vasopressin is occasionally quite useful, it hasn't proven to be a wonder drug.
Related
- EMCrit interview with Mink Chawla: Strongly recommended, with an excellent discussion focusing on the rationale for using angiotensin II. This post is intended to be considered in combination with the EMCrit podcast, to provide different perspectives. Read the ATHOS-3 manuscript, consider the pro- and con- arguments, read the Bottom Line Review of the study, and see what you think. The truth is out there.
- Vasopressor challenge concept (PulmCrit). As our menu of vasopressors expands, it becomes harder to figure out which one to choose. When in doubt, a reasonable approach is to try test doses of different agents. If the patient responds strongly to a small dose, that suggests that it's helping (e.g. if 0.03 U/min of vasopressin bumps the blood pressure a lot, that patient probably had a vasopressin deficiency). Alternatively, if the patient requires a crushingly high dose to see any measurable effect, then that drug probably isn't working very well. The ultimate goal is to support the patient's pressure and perfusion with a minimum total medication dose (noting that excessive doses of any inotrope or pressor can be toxic). Please note that this is different from the traditional approach of adding agents sequentially in a rigid fashion until the patient is on max doses of every imaginable drug simultaneously – which isn't a particularly elegant strategy.
Notes
- Full breakdown of the SOFA score over time would be required to sort this out, but I can't find it published in either the manuscript or the supplemental appendix.
- I find phenylephrine useful in patients with atrial fibrillation (doesn't speed up the heart rate) or poor IV access (minimal risk of extravasation). I have gotten some patients through septic shock with peripheral phenylephrine (patients who merely required a minimal dose of vasopressor and didn't want to get a central line. Yes, I realize this runs against the guidelines, but it's OK provided that you monitor the patient and the patient is clearly responding well to therapy).
- A recent study found that irbesartan reduced inflammation and cardiac dysfunction in a mouse sepsis model (Yousif 2017).

- Pulmcrit wee: The cutoff razor - April 15, 2024
- PulmCrit Blogitorial – Use of ECGs for management of (sub)massive PE - March 24, 2024
- PulmCrit Wee: Propofol induced eyelid opening apraxia – the struggle is real - March 20, 2024
Show me the MONEY: I agree with Josh 100%. Because this small RCT (AKA Rivers et al) was published in the NEJM folks assume that this drug must improve patient outcomes and is that it is good for patients with septic shock. No such data exists. The change (or non-change) in SOFA Score is very telling (esp when compared to the Vitamin C protocol). .
Thanks, Paul. If reduction in SOFA score, or mortality, or any other patient-centered outcome was defined as the primary endpoint of ATHOS-3, then it would have been a “negative” trial.
For the sake of comparison, see what happened to SOFA scores following ascorbic acid in Marik 2017 and Fowler 2014 here: https://emcrit.org/pulmcrit/metabolic-sepsis-resuscitation/
I am really speechless. I cannot believe that this is happening. I did a powerpoint presentation in the hospital where I work about “vasopressors”. Nothing fancy, just the evidence. And everybody was arrogantly smiling when I presented my last slide about Mr. Paul Marik “Cocktail”. Unfortunately medicine is becoming more and more a money making machine / getting the connections to do some studies and get your name published. Patients come last.
The vitamin C cocktail still does require further research to prove that it works. Several RCTs are underway currently and we should know more within the next couple of years. So it’s entirely possible that your audience was right. Currently, my opinion is that the evidence of benefit outweighs the evidence of harm. I am using the vitamin C cocktail for sepsis patients who fail to respond promptly to conventional therapy and it seems to work as reported by Dr. Marik (pressor doses decrease quickly, patients do surprisingly well). There are some conflicts of interest involving angiotenin II. For more… Read more »
It’s not really surprising the fall in cardiovascular SOFA score – considering catecholamine dose is the determinant of that score. ATII doesn’t contribute to the score, but norepinephrine does.
Agree with Steve R. The primary outcome is increase in MAp and the improved CV score in SOFA Includes the same MAP along with catecholamine vasopressors. So it’s basicall the same……In the Non placebo group, while AT2 is being used as a vasopressors, it won’t count in the SOFA score.
Agree. This is a bit murky because according the the protocol they were supposed to start weaning off the angiotensin-II at the 48-hr mark. Ideally the cardiovascular SOFA should have been obtained *after* they were entirely off angiotensin-II (because, as you and Steve point out, measuring the cardiovascular SOFA with angiotensin-II running is basically cheating). However, according to Figure S2 in the supplemental appendix, patients were *still* on Ang-II at 48 hrs so presumably they were still on the drug when the 48-hr cardiovascular SOFA was measured. I was considering mentioning this in the post but I didn’t want to… Read more »
Excellent assessment. I have long wondered why we have so little evidence about NeuroPeptide Y, Gods own norepinephrine helper. http://www.sciencedirect.com/science/article/pii/0024320594009279
Thanks. We’re only scratching the surface on septic shock – far more mediators than we understand what to do with. Unfortunately trying to sort out which ones are helping us vs. which are hurting us is no simple feat.
I agree with all of your observations or caveats, but, I would argue that you’re setting an evidentiary bar that is higher than we’d set for any other vasopressor agent. I’m unaware of ANY solid evidence that vasopressors meaningfully improve outcomes, (And there are compelling arguments that they actually kill people,) Point being that it was unrealistic to expect anything other than what we got from ATHOS-3. Whether that’s “good enough” is for each practitioner to interpret. While the concept of early multimodal therapy is conceptually interesting, I’m staying on the fence until there’s a good trial comparing it to… Read more »
any good study on pressors will use some sort of patient-centered or physiologically meaningful outcome, not soley blood pressure. for example – De Baker: NE reduced mortality vs dopa in cardiogenic shock subgroup – Ventura AM: Epi reduced mortality vs dopa in pediatric sepsis (PMID 26323041) – Numerous studies have looked at urine output, GFR, creatinine (especially studies on vasopressin). I agree with you that it’s nice to have options. The problem is that if we give La Jolla a pass here (approve and buy Ang-II on the basis of this study), then they will never do a real study… Read more »
I guess it all depends on what you define as a “meaningful endpoint”. Short term outcomes such as better urine output or higher GFR don’t impress me all that much, as I see no evidence that they meaningfully impact the outcomes that really matter. More importantly, I don’t think that these targets reflect clinical reality. When I encounter patients in refractory shock, I don’t contemplate which vasopressor cocktail is more or less likely to impact soft targets such as GFR. I use whatever works in the moment to get the MAP high enough to keep organs perfused and the patient… Read more »
We should look at this however and say what’s the cost… it’s going to cost $1500 for a single use vial… if you have two therapies without strong data to direct you – cost should a’at least’ come into play. Marik’s cocktail is much lower cost than ATII. Ton of potential industry bias in the ATHOS-3 trial, PI works for the company, bias bias bias $$$$$, weak data.
I’d add that the patient/family/system often has ‘something to lose’ when we are ‘throwing a hail mary’.
Well, duh. If the patient is dying, he/she by definition has everything to lose. And the worse the situation, the more likely we are to start reaching for whatever might work.
I submit that any intensivist who hasn’t occasionally tried something at least a little crazy to save a dying patient is either a liar or a crappy intensivist.
That comment was meant more as a end of life comment – I was more talking about saving someone that is that close to death – the outcome in the short and long term is not likely to be good if they are on that end of the spectrum of septic shock. The family gets false hope, etc. The system gets minimal/no return on it’s investment, lost opportunity cost. This medication is ‘far’ more expensive than epi/norEpi at supraphys doses, Marik’s cocktail, outside of bridging a pt from profound pump vasoplegia I don’t use methylene blue – before we spend… Read more »
IV vit C is actually pretty expensive, around $4000 for the vit c protocol. Don’t think either has enough data but AT ii in patients requiring CRRT might be worth it based on the few patients if it reduces time on CRRT
if you’re paying that much for VitC you’re being robbed, the cost should be at least 10x lower.
I’m surprised to not see a cost discussion here as well. When I inquired how much the price was to order at my institution I was told about $1000 for a 1mL vial. Cost isn’t even in the ball park of the numerous pressors we already stock. I too was shocked to see that the FDA pushed this through. And now it feels like providers feel obliged to pursue. With what is available already on the market we typically are able to meet MAP goals. If they were able to show improved mortality or less atrial fibrillation I may have… Read more »
What do you think of this?
https://www.beckershospitalreview.com/quality/tackling-sepsis-how-innovative-therapies-and-treatment-protocol-can-save-lives.html
How about this? Giapreza seems to be efficacious and just won medicare approval as baing a drug superior to others in it’s class. Perhaps an update to this article is in order?
https://youtu.be/o8QPSV2WFX4
Cedric Franklin Rutland I love your site/posts. They are all thought provoking. This article is just as thought provoking. As you mentioned, blocking the RAS in animal models improves outcomes…I just read a study in the British journal of pharmacology stating that using an ARB to block may improve outcomes but using an ACE actually increases risk of mortality in septic shock. Again, all of this is interesting which is why we do our jobs. Our bodies are not black and white. Many proteins serve different functions at different times in different areas of the body. The fact is we… Read more »
While I share the complete lack of enthusiasm for the new agent and the poorly manufactured study that gained its FDA approval, I do believe for arguments sake a number of these comments may be misleading. 1. To be fair there is more significant evidence and well designed trials suggesting a benefit of catecholamine blockade or augmentation (beta-blockers, clonidine, dexmedetomidine) in septic shock so this theory/controversy exists universally and in much better evidence then animal models. (https://www.ncbi.nlm.nih.gov/pubmed/24108526, https://www.ncbi.nlm.nih.gov/pubmed/26081003). Again I would suggest that giving catecholamines, as we do in common practice, in a catecholamine toxic state such as septic shock… Read more »
I haven’t been able to find any preliminary/presented or published data on the angiotensin II blocker septic shock study (https://clinicaltrials.gov/ct2/show/NCT01992796). Info was last updated in 2013. Enrollment was to be completed by 2017. No results are posted. It’s going on 2 years with no info. Wonder what happened to the trial??
excellent Josh… as always.
I was very hopeful after the interview with Dr Chawla,.. we shall see.
This article needs to be updated as more and more evidence is piling up showing the benefits of this vs other Vasopressors. Also with drugs like Vasopressin skyrocketing their prices ATII is beginning to look much more attractive. Not to mention, IT’S LITERALLY SAVING LIVES.
I would love to see you provide an update on this. Have had some great experience with Ang2 working well in the right patient used early.