

CONTENTS
- Thrombotic Microangiopathy(TMA)
- Thrombotic thrombocytopenic purpura (TTP)
- Shiga-Toxin mediated Hemolytic Uremic Syndrome (ST-HUS)
- Complement-mediated Hemolytic Uremic Syndrome (C-HUS)
- TMA in pregnancy or postpartum
- Podcast
- Questions & discussion
- Pitfalls

schistocytes

- Schistocytes are fragmented erythrocytes which result from intravascular hemolysis (figure above).
- Schistocytes can occasionally be seen in normal patients, but at very low concentrations (<<0.5% of erythrocytes).
- The presence of >0.5% schistocytes suggests a microangiopathic hemolytic anemia, which is defined as…
microangiopathic hemolytic anemia (MAHA)
- MAHA refers to non-immune intravascular hemolysis – essentially erythrocytes being torn apart within the bloodstream.
- Clinically, MAHA is revealed by the combination of hemolytic anemia plus detection of schistocytes on the blood smear.
- 💡 Intravascular hemolysis causes hemoglobinuria, which may be suspected if the urine is heme-positive without the presence of erythrocytes.
- Other laboratory abnormalities of hemolytic anemia should be present as well (figure below).
- Causes of MAHA include:
- (1) Physical fragmentation of erythrocytes by prosthetic valves or mechanical devices.
- (2) Thrombotic microangiopathies, which are…
thrombotic microangiopathy (TMA)
- TMA refers to a variety of conditions marked by the development of tiny clots in the microvasculature.
- Clinically, TMAs generally present as a combination of three hallmark features:
- (1) MAHA (revealed by hemolytic anemia plus schistocytes) – erythrocytes rip open as they are forced through small blood vessels which are partially occluded.
- (2) Thrombocytopenia – either an absolute platelet count <150,000 or a 25% reduction from baseline)(31935318)
- (3) Ischemic tissue damage due to microvascular occlusion. Organs involved may include the kidney, brain, heart, lungs, gut, or skin. Different microangiopathies tend to involve different organs.

primary thrombotic microangiopathies
- Thrombotic thrombocytopenic purpura (TTP)
- Inherited thrombotic thrombocytopenic purpura (congenital deficiency of ADAMTS13).
- Acquired thrombotic thrombocytopenic purpura (antibody impairs function of ADAMTS13).
- Shiga Toxin related Hemolytic Uremic Syndrome (ST-HUS) – this follows a diarrheal illness in 95% of cases, with organ damage often limited to the kidney. This is also known as “typical HUS.”
- Complement mediated HUS (C-HUS) – this often follows infection or pregnancy, with more widespread organ damage than in ST-HUS. This is also known as “atypical HUS.”
TMA-associated conditions (a.k.a., “secondary TMA”)
- TMA may also be caused by numerous disorders as listed below (e.g., medications and infections). This is sometimes termed “secondary TMA,” but the term “secondary TMA” is often misleading:
- (1) Most patients exposed to these conditions will not develop a TMA.
- (2) In many cases, the conditions listed below will trigger a primary TMA (e.g., infection or pregnancy triggering C-HUS).
- Thus, there is often overlap between primary and secondary TMA – wherein a triggering condition leads to an episode of TTP or especially C-HUS. Therefore, finding a cause of “secondary” TMA doesn't exclude the occurrence of a “primary” TMA. Instead, the possibility of underlying TTP or C-HUS should always be considered.(29582550)
- Episodes of TMA may result from a combination of the patient's underlying tendency to develop TMA (e.g., complement system gene mutations) plus the intensity of the triggering cause.
- Some authors have suggested that the majority of secondary TMA actually represents C-HUS.(30865166)
TMA associated with rheumatologic disease
- SLE.
- Scleroderma (including scleroderma renal crisis).
- Antiphospholipid antibody syndrome (including Catastrophic Antiphospholipid Syndrome).
TMA associated with pregnancy – explored below.
TMA secondary to malignant hypertension
- It may be difficult to sort out if hypertension is causing TMA, or whether a primary TMA is causing hypertension (as a result of renal involvement). C-HUS, glomerulonephritis, or scleroderma renal crisis in particular may cause severe hypertension.(32539032) Clues which may help sort this out:
- If known, the sequence of events may be helpful.
- Absence of a history of severe hypertension may suggest the presence of a TMA causing hypertension.
- Severe thrombocytopenia may favor the presence of TMA, rather than solely hypertension.(32539032)
- Failure to improve after controlling the blood pressure excludes hypertension as the sole disease process.
drug-induced TMA (33841853)
- Quinine* (including tonic water 🍸).
- Antiplatelets (clopidogrel, ticlopidine, prasugrel).
- Immunosuppressives:
- Calcineurin inhibitors (cyclosporine, tacrolimus).
- m-TOR inhibitors (sirolimus, everolimus).
- Oncologics:
- Mitomycin C, gemcitabine*, oxaliplatin.*
- VEGF inhibitors (e.g., bevacizumab, sunitinib, pazopanib).
- Proteasome inhibitors (bortezomib, carfilzomib).
- BCR-ABL inhibitor imatinib.
- Antimicrobials (acyclovir, ciprofloxacin, metronidazole, nitrofurantoin, trimethoprim/sulfamethoxazole).
- Miscellaneous:
- Estrogen/progesterone.
- Quetiapine.*
- NSAIDs (ibuprofen, ketorolac).
- Type-I Interferon.
- Oxymorphone (Opana), intravenous misuse of oral opioids.(32259874)
- Simvastatin.
- Cocaine.
- Qualifiers: Drug-associated TMA may be acute or chronic. *Indicates medications which may cause immune-mediated TMA, which can present explosively.
infection-associated TMA
- Bacterial endocarditis.
- Streptococcus pneumoniae.
- Rocky Mountain Spotted Fever (RMSF).
- Erythrocyte parasites (malaria, babesia).
- Systemic aspergillosis.
- HIV.
- CMV, EBV.
- Influenza H1N1.
glomerular diseases associated with TMA
- IgA nephropathy.
- ANCA-associated vasculitis.
malignancy-associated TMA
- Systemic malignancy with microvascular obstruction by tumor cells (mostly: gastric, breast, prostate, lung).(32950988)
- Due to chemotherapy:
- (1) Chemotherapeutic medications listed above.
- (2) Regimens used for bone marrow ablation, prior to hematopoietic stem cell transplantation.
transplantation-associated TMA
- Acute rejection of a transplanted kidney.
- Calcineurin toxicity in solid-organ transplant recipients.
- Recurrence of C-HUS in renal transplant patients (if renal failure was initially due to occult C-HUS).
- Treatment regimens for graft versus host disease (GVHD).
- Myeloablative chemotherapy prior to bone marrow transplantation.
- Opportunistic infections associated with transplantation (e.g., CMV).
non-TMA entities to consider on the differential diagnosis
- Mechanical erythrocyte fragmentation by prosthetic valve, cardiac assist devices, or transjugular intrahepatic portosystemic shunts (TIPS).
- Severe B12 deficiency (may rarely cause thrombocytopenia & microangiopathic hemolytic anemia).
lab panel to evaluate the cause of a thrombotic microangiopathy
- MAHA labs:
- Manual blood smear.
- LDH (lactate dehydrogenase).
- Haptoglobin.
- DIC labs (INR, PTT, fibrinogen, D-dimer).
- CBC with differential.
- Liver function tests (including indirect bilirubin).
- Troponin and EKG.
- Urinalysis & microscopic examination of the urine sediment.
- Pregnancy testing as appropriate.
- Infectious workup:
- Blood cultures.
- Other relevant infectious evaluation (e.g., HIV testing, pneumococcal urinary antigen).
- ADAMTS13 antigen level & anti-ADAMTS13 antibodies or inhibitor (depending on which tests are available).
- Antinuclear antibodies (ANA).
- Stool PCR analysis for Shiga-toxin genes, stool culture, and/or bioassay of stool for Shiga toxin (depending on the local availability of these tests).
- Other studies to evaluate for an underlying cause will depend on clinical context, for example:
- If B12 abnormality is suspected: obtain serum homocysteine and methylmalonic acid levels.
- If catastrophic antiphospholipid antibody syndrome (CAPS) is suspected: anticardiolipin antibody (IgG & IgM), anti-beta-2-glycoprotein type I (IgG & IgM), and dilute Russell viper venom time (more on evaluation of CAPS here).
interpretation of labs to elucidate the etiology of TMA
complete blood count
- Thrombocytopenia: In TTP, thrombocytopenia is often severe. In one series, only 4% of TTP patients had a platelet count above 30,000.(29296701) Thus, platelet counts <30,000 favors TTP.
- Markedly elevated MCV: Consider B12 deficiency.
coagulation labs
- INR, PTT, and fibrinogen are generally normal in most thrombotic microangiopathies (e.g., INR <1.5).
- If DIC is present, this suggests the presence of another underlying process (e.g., sepsis). However, TTP can occasionally cause DIC due to extensive tissue necrosis.
- Diagnostic criteria for DIC: 📖
- Isolated elevation of PTT may raise a question of lupus anticoagulant.
creatinine level
- TTP:
- TTP causes acute kidney injury in 50% of patients, but this is generally mild.
- Mild or absent renal involvement would favor TTP.
- Substantial acute renal failure (creatinine >2-2.3 mg/dL) argues against TTP.
- ST-HUS or C-HUS may cause gradual development of severe renal failure over several days.
- Drug-induced TMA may cause extremely rapid renal failure, with abrupt-onset anuria.
urinalysis
- Muddy brown casts may suggest acute tubular necrosis, which would tend to imply a non-TMA cause of renal failure.
- Red blood cell casts would imply the presence of glomerulonephritis, which could focus the differential diagnosis on TMA due to glomerulonephritis (listed above).
ADAMTS13 tests
- ADAMTS13 activity
- Undetectable levels (<10% normal levels) are seen in almost all TTP patients. However, this isn't always 100% specific for TTP, as it can also be caused by some infections or malignancies.
- Low activity (10-60% of normal levels) is commonly seen among ill patients with systemic inflammation (e.g., malignancy or sepsis) or hemolytic uremic syndrome.(33841853) Some TTP patients can have levels at the lower end of this range (e.g., ~10-20%), especially if they received plasma prior to measurement of ADAMTS13 level.
- Normal levels (>60% of normal) exclude TTP.
- ADAMTS13 inhibitor
- Most acquired TTP is due to antibodies which block the activity of ADAMTS13. Such antibodies may be detected as the presence of an ADAMTS13 inhibitor in mixing studies (i.e., mixing the patient's plasma with normal plasma causes an inhibition of ADAMTS13 activity in the normal plasma).
- Acquired TTP may not reveal measurable inhibitor activity in some situations (e.g., an antibody which doesn't inhibit ADAMTS13 enzymatic activity directly, but rather causes accelerated clearance of ADAMTS13 from the blood).
- Hereditary TTP is due to inadequate synthesis of ADAMTS13, so patients with hereditary TTP won't have an ADAMTS13 inhibitor.
- ELISA assay for antibodies which bind to ADAMTS13
- This tests for any antibodies binding to ADAMTS13 (whether or not they are neutralizing).
- This test increases the sensitivity for detection of acquired TTP, at the cost of some reduction in specificity.
- ☎ Assays will vary between hospitals, so discuss with the lab which tests to order.
antinuclear antibodies (ANA)
- A positive ANA is generally interpreted to favor a diagnosis of TTP in the context of thrombotic microangiopathy, since roughly half of TTP patients will have a positive ANA.(30294946) Some scoring systems have formally incorporated the use of ANA as an indicator of TTP.(30504354)
- Of course, ANA isn't specific for any disease. ANA is often positive in patients with catastrophic antiphospholipid antibody syndrome (CAPS), so this should also be considered.(more on CAPS here)

initial treatment of TMA is a paradox
- TTP requires emergent therapy with plasmapheresis.
- C-HUS requires urgent treatment with eculizumab.
- ST-HUS requires only supportive therapy.
- Definitive diagnosis requires laboratory tests which often take several days to return (e.g., ADAMTS13 labs).
Therefore, empiric therapy must often precede definitive diagnosis. Clinical judgement is required to rapidly assess the likelihood of TTP or C-HUS. As laboratory data returns, the diagnosis and treatment may change (e.g., an initial empiric diagnosis of TTP may be changed to a diagnosis of probable C-HUS, after a normal ADAMTS13 level is reported).
empiric treatment for TTP with plasmapheresis and steroid
TTP is usually the initial consideration
- TTP is a medical emergency for which prompt plasmapheresis reduces mortality. Therefore, plasmapheresis is often empirically initiated before the diagnosis of TTP is definite.
- Among all the thrombotic microangiopathies, TTP may be the greatest life threat (due to its frequent involvement of the heart and brain).
- Steroid should be considered in combination with plasmapheresis when treating TTP empirically. 📖
PLASMIC score
- The PLASMIC score may help determine the likelihood that the patient has TTP and, thus, the benefit of plasmapheresis:
- 🛑 Note that the PLASMIC score is applicable only to patients with schistocytes, thrombocytopenia, and a clinical question of possible TTP.
- The score ranges from 0-7, with one point assigned for each of the following. The earliest labs available should be used to determine the score.
- Platelet count <30,000.
- Hemolysis: either reticulocyte count >2.5%, haptoglobin undetectable, or indirect bilirubin >2 mg/dL (>34 uM/L).
- No active malignancy within one year.
- No history of transplantation.
- MCV <90 fL.
- INR <1.5.
- Creatinine <2 mg/dL (177 uM). (⚠️ Careful however – if the baseline creatinine is elevated, this criterion may not be valid.)
- A score of 5-7 has a high sensitivity for TTP, ~99% in some series.(32757237) Scores 5-7 suggest a benefit from empiric plasmapheresis, unless there is an alternative diagnosis which explains the patient's abnormalities.
- Scores of 0-4 suggest that TTP is unlikely, and that further evaluation for TTP is generally unnecessary.
the French score 🇫🇷
- This is an alternative scoring system that is fundamentally quite similar to the PLASMIC score (table below).(32914582) It may be a bit easier to use in some situations. Note, however, that the FRENCH score is only applicable to a smaller group of patients, those with:
- Thrombocytopenia.
- Significant microangiopathic hemolytic anemia (e.g., substantial schistocytes).
- No history or evidence of cancer.
- No prior transplantation.
- Absence of DIC.
- ⚠️ Older patients may have higher baseline creatinine values, causing false-negative results from these predictive scores. In one study, all false-negative results utilizing the French score occurred among patients >55 years old.(32259874) A similar phenomenon has been reported with the PLASMIC score.(33179792) Thus, TTP cannot be excluded on the basis of elevated creatinine in older patients.

empiric eculizumab for C-HUS
- The criteria to initiate empiric eculizumab for C-HUS are less well-defined. Factors that might favor initiation of eculizumab include the following:
- TMA with a low index of suspicion for TTP or ST-HUS.
- Lack of any alternative explanation for TMA (other than C-HUS).
- Severe renal involvement (the rationale for early eculizumab is to salvage renal function).
- A compatible clinical context suggestive of C-HUS (especially postpartum TMA).
- The diagnosis of C-HUS is often not initially evident, so it may become a diagnosis of exclusion.
- More on the diagnosis and treatment of C-HUS below.


TTP results from the following sequence of events
- (#1) The underlying problem is a deficiency of ADAMTS13, the enzyme that degrades multimers of von Willebrand factor.
- a) Usually this deficiency is due to an autoantibody against ADAMTS13 (acquired TTP).
- b) Rarely this is due to a congenital deficiency in ADAMTS13 production (congenital TTP).
- (#2) Huge multimers of von Willebrand factor accumulate.
- (#3) Multimers cause platelet agglutination in small blood vessels, causing microthrombus formation.
- (#4) Microvascular occlusion from platelet agglutination causes shearing of erythrocytes, leading to microangiopathic hemolytic anemia. Vascular occlusion may also cause tissue damage.
congenital TTP (a.k.a., Upshaw-Schulman syndrome)
- Congenital TTP is an autosomal recessive disorder caused by inadequate ADAMTS13 activity.
- Congenital TTP can present in adulthood, especially in the context of pregnancy. Among adults, congenital TTP accounts for ~10% of TTP cases.
- Treatment of congenital TTP is generally similar to the treatment of acquired TTP. However, there is no role for immunosuppression (since patients do not have pathological autoantibodies). Since these patients lack anti-ADAMTS13 antibodies, simple administration of plasma may be more effective (without necessarily requiring plasmapheresis).
- The remainder of the discussion below will focus on acquired TTP, as this accounts for 90% of cases among adults.(33540569)
- Primarily seen in young adults (~20-50 years old).
- Two-fold female predominance.
- Eight-fold increased rate among African Americans.(33540569)
- TTP may be caused by certain conditions (“secondary TTP”):
- Pregnancy (more on this below).
- Rheumatologic disease (mostly SLE).
- Less often: Malignancy, HIV, ticlopidine.
the classic pentad of TTP:
- ⬟ Thrombocytopenia (100%).
- Often with purpura.
- However, patients usually don't have severe bleeding.
- ⬟ Microangiopathic hemolytic anemia (100%).
- ⬟ Neurologic symptoms (~70% initially):
- Symptoms often rapidly wax and wane.
- Wide variety of symptoms may occur: headache, seizure, stroke or transient focal abnormalities, delirium, coma, posterior reversible encephalopathy syndrome (PRES).
- Note that with aggressive therapy complete neurologic recovery is generally seen. Therefore, profound neurologic impairment shouldn't dissuade aggressive therapy.
- ⬟ Kidney injury (~50% initially):
- Renal failure is generally milder in TTP than in other types of TMA.
- With current management strategies, it's highly unusual for patients to progress to fulminant renal failure.
- ⬟ Fever (~20% initially).
- 💡 The classic pentad is present only in ~5% of patients, so this is not useful as a diagnostic criterion.
other symptoms:
- Gastrointestinal symptoms occur in roughly half of patients:
- Common symptoms include pain, nausea/vomiting, and diarrhea.
- Pancreatitis can occur.
- Bloody diarrhea can occur due to mesenteric microthrombosis, causing colitis. This may cause some diagnostic confusion with regards to Shiga toxin-mediated HUS. Unlike in ST-HUS, patients with TTP experience GI symptoms simultaneously with microangiopathic hemolytic anemia.
- Cardiac involvement:
- Myocardial infarction.
- Acute heart failure.
- Arrhythmias.
(#1) Fresh frozen plasma infusion while waiting for plasma exchange
- Fresh frozen plasma contains ADAMTS13, so this may be beneficial.
- If plasma exchange is going to be delayed (e.g., >6-8 hours), it may be helpful to give 2 units of fresh frozen plasma, followed by ~1 unit every four hours. Consider administration of a diuretic along with plasma, to avoid volume overload.
(#2) plasmapheresis (a.k.a., plasma exchange)
- This is the mainstay of therapy. Plasmapheresis works via removal of vWF multimers, removal of anti-ADAMSTS13 antibody, and addition of ADAMTS13 enzyme.
- Plasma exchange should be initiated promptly, ideally within <6 hours after the diagnosis of thrombotic microangiopathy.
- Technical details of plasmapheresis:
- (1) DO NOT give platelets to “facilitate” placement of a hemodialysis catheter for plasmapheresis. Dialysis catheter placement in TTP appears to be safe regardless of the platelet count.(31588978) The catheter should be inserted carefully, using ultrasound guidance, by an expert operator.
- (2) Meticulous sterility is critical when placing these catheters, as they will often remain in place for over a week.
- (3) Hypocalcemia may arise during plasma exchange.(more on hypocalcemia here).
- (4) Consider scheduling administration of medications after plasmapheresis sessions, to avoid excessive medication clearance.(31588978)
- (5) Make sure to send all pertinent labs prior to plasmapheresis, as many labs may be altered by the procedure.(see lab panel above)
- Patients should receive daily exchanges of 60 ml/kg (1.5 plasma volumes). However, in patients with life-threatening neurological or cardiac involvement, plasmapheresis may be performed twice daily.(26418759)
- Plasmapheresis is usually continued until the platelet counts remain >150,000/mm3 for two consecutive days. Subsequently, plasmapheresis may be discontinued (with close clinical monitoring). Lactate dehydrogenase levels may also be used to gauge the response to plasmapheresis.(31588978)
(#3) immunosuppressive therapy
- Steroid
- In adults, TTP is generally due to an acquired antibody against ADAMTS13, which may respond to steroid.
- One RCT suggested that starting with high-dose steroid could improve remission.(20033409) This study used 10 mg/kg/day methylprednisolone for three days, then 2.5 mg/kg/day.
- An alternative regimen is 1 mg/kg/day oral prednisone.(29582550), This may be a more sensible dose for patients in whom the diagnosis of TTP is not definite, or for patients with milder disease.(31588978)
- Rituximab
- Rituximab has not been demonstrated to be beneficial within a randomized controlled trial.
- 🛑 Rituximab may render patients vulnerable to chronic, untreatable COVID infection. Currently, this treatment should be withheld when possible.
(#4) blood pressure control
- Correlational studies suggest that hypertension may aggravate erythrocyte fragmentation and worsen the disease process.
- Hypertension should probably be controlled, with a target systolic blood pressure of perhaps <140 mm.(22292070)
(#5) avoid blood products (other than fresh frozen plasma)
- Platelets:
- Giving platelets may worsen thrombosis and tissue ischemia. Therefore, platelet administration should be avoided unless there is a clinically significant hemorrhage.
- 🛑 Don't give platelets prior to placement of a plasmapheresis catheter (as discussed above).
- Red blood cell transfusion:
- Avoid overtransfusion, which may simply aggravate hemolysis.
- Consider transfusion only if hemoglobin is <7 mg/dL and this is causing symptoms.
- Folate should be supplemented to promote endogenous hematopoiesis.(31588978)
(#6) Venous thromboembolism prophylaxis
- Patients are at high risk of venous thromboembolism. The presence of thrombocytopenia is not protective against this.
- Prophylactic heparin should be started early (e.g., when platelet count rises above ~30,000/mm3).
(#7) antiplatelet therapy
- It appears safe and reasonable to provide low-dose aspirin to patients with a platelet count >50 billion/L.(29582550; 9299856). Aspirin may be especially reasonable among patients with myocardial infarction (see below).
- Among patients who are receiving caplacizumab, the safety of aspirin is unknown.
(#8) myocardial infarction
- Pathophysiology: This is a common manifestation of TTP due to microvascular disease. Epicardial coronary artery occlusion is extremely rare.
- Potentially useful treatments:
- (1) Aggressive treatment for TTP (e.g., twice daily plasmapheresis).
- (2) Aspirin is indicated for patients with a platelet count >50, as it generally is (see #7 above).(26418759)
- (3) Blood pressure control with beta-blockers may reduce myocardial workload (assuming that the patient isn't in acute heart failure).(30622684)
- Treatments that are not generally helpful:
- Usually these patients don't have a plaque rupture, so typical treatment for MI may not work well.
- Cardiac catheterization is generally inadvisable for several reasons: Catheterization may worsen kidney injury, the stent may clot off, and placing a stent commits the patient to dual antiplatelet therapy (which may be problematic in the context of fluctuating platelet counts).(30622684)
- Vasopressin should be avoided, as this may cause secretion of von Willebrand factor, thereby exacerbating TTP.
(#9) caplacizumab
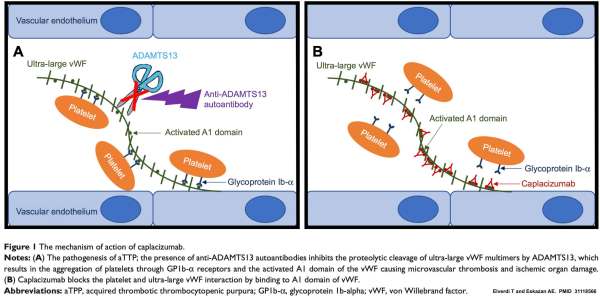
- Caplacizumab is an antibody fragment that binds to von Willebrand factor (vWF). It blocks the interaction between von Willebrand factor and platelets, thereby preventing the formation of microthrombi (figure above). This offers the ability to immediately reduce thrombus formation (even before the level of ADAMTS13 can be restored).
- Caplacizumab should be considered, especially for more severe situations (e.g., neurologic or cardiac involvement). The primary drawback of caplacizumab is an increased risk of mild bleeding. The HERCULES RCT found that caplacizumab reduced the mean number of ICU days from ten to three.
- ISTH guidelines recommend the use of caplacizumab.(32914526)
- Caplacizumab may cause clinical improvement even despite a low level of ADAMTS13. This creates a risk of disease relapse after caplacizumab is discontinued, if the underlying ADAMTS13 deficiency hasn't been adequately addressed.
- Shiga-toxin related Hemolytic Uremic Syndrome (ST-HUS) is caused by Shiga toxins that are produced by infection with Shigella or certain strains of E. coli (E. coli 0157:H7 and O104:H4). Toxins enter the body and damage endothelial cells, leading to the formation of microthrombi.
- ST-HUS is often referred to as “typical” HUS since it is more common (especially in children). The terminology “ST-HUS” is used here because it is more pathophysiologically accurate and because ST-HUS is more “typical” only in some clinical contexts.
(#1) exposure
- Patients may report exposure to raw meat, animals, contaminated water, or unpasteurized milk. Exposure occurs several days before any symptoms occur.
- Occasional outbreaks occur, but most cases are sporadic.
(#2) gastroenteritis
- The initial clinical presentation is with a diarrheal illness (e.g., abdominal pain, nausea/vomiting, and diarrhea). However, clinical colitis may be absent in 5% of patients, so absence of a diarrheal illness doesn't exclude ST-HUS.(32539032)
- Diarrhea often becomes bloody, which may trigger inappropriate concerns regarding gastrointestinal hemorrhage.
- Differential diagnosis: gastrointestinal symptoms can also be caused by bowel ischemia as a result of TMA. The key to sorting this out may be timing: ST-HUS is suggested if gastrointestinal symptoms precede renal dysfunction by some days.
(#3) thrombotic microangiopathy
- Several days after gastrointestinal symptoms, TMA occurs with thrombocytopenia.
- The primary organ involved is the kidney. Acute kidney injury is often severe.
- Neurologic involvement may occur as well (with symptoms including altered mental status, focal abnormalities, seizure, or coma).(32172817)
assays for ST-HUS
- Roughly three types of assays are available:
- Traditional stool cultures.
- PCR analysis of the stool for the Shiga-toxin gene.
- Bioassays of stool for the presence of Shiga toxin.
- The availability of various assays will vary across hospitals. Confer with your laboratory to determine the optimal approach.
clinical value of a definitive diagnosis of ST-HUS
- Unfortunately, there is no specific therapy for ST-HUS. However, a definitive diagnosis is useful, since this allows for confidently avoiding plasmapheresis or eculizumab.
- ST-HUS is a reportable disease. Notification of public health services may assist in source tracking and preventing further exposures of other people.
- There is no high-quality evidence that plasmapheresis is beneficial for ST-HUS. Therefore, plasmapheresis is generally not recommended for ST-HUS.
- Treatment is essentially supportive.
- In patients with volume depletion due to diarrhea, this should be corrected with crystalloid resuscitation.
- Hypertension may require management with antihypertensives.
- Hemodialysis is frequently required in the acute disease phase, but renal recovery often occurs such that long-term hemodialysis may not be required.
- 🛑 Platelet transfusion should be avoided unless truly necessary (e.g., clinically significant bleeding), since platelet transfusion may exacerbate microthrombosis.
basics
- Complement-related Hemolytic Uremic Syndrome (C-HUS) is due to hyperactivity of the atypical complement system.
- C-HUS is commonly referred to as “atypical HUS” (aHUS). The term C-HUS is used here because it is more pathophysiologically accurate, and because C-HUS might actually be more common than we realize.
- C-HUS requires specific therapy (eculizumab), so early consideration and diagnosis is important.
pathophysiology

- C-HUS results from uncontrolled activation of the alternative complement system. Causes of this dysregulation may include:
- i) Genetic deficiency of the regulatory proteins that normally reduce activation of the alternative pathway of complement (e.g., complement factor H, membrane cofactor protein). Although these abnormalities are hereditary, C-HUS commonly presents in adulthood.
- ii) Acquired deficiency of complement factor H or complement factor I, caused by autoantibodies.
- Following a trigger, inadequate complement regulation may lead to rampant alternative complement activation that damages the vascular endothelium. Endothelial damage causes microvascular thrombosis, leading to a thrombotic microangiopathy.
- Systemic release of anaphylatoxins C3a and C5a generated in the kidney may lead to histamine release, causing interstitial edema.(30145224)
(#0) rarely, a history of C-HUS
- If you're lucky, a family or a personal history of a complement abnormality might be present.
- However, most cases are sporadic, rather than familial – so a history will be absent.(29582550)
(#1) preceding trigger
- Infection is involved in ~50% of adult cases (including gastroenteritis, viral illnesses, and sepsis).
- Medication or substance exposure (e.g., bleomycin, cisplatin, mitomycin-C, quinine, cocaine).
- Surgery.
- Pregnancy is a classic trigger for C-HUS (more on this below).
(#2) thrombotic microangiopathy
- Renal dysfunction is often present.
- This is the hallmark organ failure, potentially requiring hemodialysis.
- Hematuria and proteinuria are common, with proteinuria occasionally increasing into the nephrotic range.(30294946)
- Hypertension
- Extrarenal manifestations are seen in ~20% of patients. Catastrophic presentation with multiorgan failure can occur in 5% of patients (30145224; 30294946)
- CNS involvement is the most common extrarenal manifestation. Features may include headache, confusion, focal neurological abnormalities, seizure, or coma. Imaging may show symmetric involvement resembling posterior reversible encephalopathy syndrome (PRES).
- Cardiac involvement occurs in ~10% of patients. This may include myocardial ischemia, arrhythmias, myocarditis, and reduced ejection fraction.(30031798)
- Visual changes may occur, reflective of exudative or ischemic retinopathy.
- Respiratory failure may result from alveolar edema, pleural effusions, or pulmonary hemorrhage.
- Gastrointestinal manifestations may include pancreatitis, hepatitis, or colitis (including intestinal bleeding, obstruction, and perforation).

laboratory diagnostics are inadequate
- Currently most hospitals lack any complement testing capable of diagnosing C-HUS in a timely and accurate fashion.
- C3, C4, and CH50 may be measured rapidly, but these tests lack adequate sensitivity and specificity.
- More sophisticated assays (e.g., soluble C5b-9 levels) might be useful, but these are not widely available.
- Mayo Clinic offers a C-HUS screening panel, but this requires up to 21 days to return results. Sending this test prior to initiation of eculizumab may be considered, but the test won't return rapidly enough to affect ICU management.
- Genetic testing for C-HUS takes weeks. Roughly half of patients with C-HUS lack detectable genetic abnormalities (many of which haven't been discovered yet). To complicate matters further, many detectable genetic anomalies have unclear clinical significance.
diagnostic approach to C-HUS
- Currently, C-HUS diagnosis in the context of critical illness remains a matter of clinical judgement. Key considerations include:
- (i) The presence of a thrombotic microangiopathy which predominantly involves the kidneys.
- (ii) Either exclusion or improbability of TTP or ST-HUS.
- (iii) Lack of an alternative explanation for the patient's thrombotic microangiopathy.
- (iv) Clinical scenario suggestive of C-HUS (especially postpartum renal failure).
- In some situations it may be reasonable to reach a presumed diagnosis of C-HUS and initiate eculizumab, before the results of all diagnostic and genetic testing are returned.
- Plasmapheresis is not generally effective.
- Depending on the precise mechanism of complement dysregulation, plasmapheresis could theoretically be beneficial in some cases.
- Plasmapheresis is not generally recommended for C-HUS, given that eculizumab should be uniformly effective (regardless of the precise mechanism underlying the patient's C-HUS).
- Causative factors should be controlled, if possible.
- Eculizumab is definitive therapy. This is a monoclonal antibody fragment that inhibits C5 convertase, thereby blocking the terminal complement pathway (figure below).
- Eculizumab may cause renal recovery even in patients where the patient has progressed to hemodialysis.
- The standard initial adult dosing is 900 mg weekly for four weeks.
- Eculizumab increases the risk of infection by encapsulated bacteria (especially Meningococcus spp.), so patients should be vaccinated against encapsulated pathogens. Until immunity develops, empiric antibiotic coverage might be a consideration (e.g., penicillin V potassium).(30031798)
- Platelet transfusion should be avoided unless truly necessary (e.g., clinically significant bleeding), since platelet transfusion may exacerbate microthrombosis.
diagnosis of TMA in pregnancy or postpartum
- This is overall similar to the diagnosis of TMA in general. However, in pregnancy, thrombocytopenia may be redefined as <100 platelets (since platelets normally fall during pregnancy)(32808006)
differential diagnosis

- Preeclampsia, eclampsia, and HELLP syndrome (more on this here).
- HELLP syndrome is a TMA affecting mostly the liver and more rarely the kidney.
- Epidemiologically, these conditions are generally more common than other forms of thrombotic microangiopathy.
- HELLP syndrome is strictly related to pregnancy, so it should resolve rapidly following delivery.(29348275)
- TTP
- The normal physiology of pregnancy involves an increase in von Willebrand factor levels and a reduction in ADAMTS13 levels.(29348275) Pregnancy may thus trigger either acquired (autoimmune) TTP, or congenital TTP.
- TTP tends to occur in second or third trimester.
- C-HUS
- This is the only form of TMA to occur most often in the postpartum period (up to three months after delivery). However, C-HUS can occur during any trimester.
- TMA beginning in the postpartum period after an uneventful pregnancy is very suggestive of C-HUS.(32808006)
- SLE flare
- Catastrophic antiphospholipid antibody syndrome (more on this here).
management

- Expedited delivery may be required in some situations (particularly preeclampsia/HELLP), but delivery doesn't seem to have a substantive effect on other pathologies (e.g., TTP or C-HUS). The timing and mode of delivery will be determined by the obstetrics team.
- TTP: For patients in whom TTP is likely, empiric plasmapheresis and steroid therapy may be appropriate (while awaiting additional data).
- Preeclampsia/HELLP:
- Between 20 weeks gestation and the early postpartum period, preeclampsia/HELLP is the most common cause of TMA. If patients meet the criteria for preeclampsia/HELLP, this is the most likely diagnosis and it should be treated accordingly (more on this here).
- Emerging evidence suggests that HELLP may involve dysregulation of the complement system, similar to C-HUS. Thus, eculizumab may be considered as adjunctive therapy for some patients with HELLP who have prominent features of thrombotic microangiopathy.(30865166)
- C-HUS:
- Eculizumab is the treatment of choice for C-HUS in pregnancy. Higher doses may be required than usual, due to higher volume of distribution and/or urinary loss in patients with heavy proteinuria.(32808006)
- In some patients with TMA occurring several weeks postpartum and primarily involving the kidney, empiric initiation of eculizumab may be reasonable (since this presentation is strongly suggestive of C-HUS).

Follow us on iTunes
To keep this page small and fast, questions & discussion about this post can be found on another page here.

- TTP is a medical emergency which often requires aggressive empiric therapy before definitive diagnosis. If you're considering this diagnosis, consult hematology immediately to determine if urgent plasmapheresis is needed.
- Platelet transfusion is contraindicated (may worsen thrombosis).
- Standard therapies for myocardial infarction will generally fail in patients with thrombotic microangiopathy, and may actually exacerbate matters (myocardial ischemia is due to micro-thrombosis, not plaque rupture).
- Make sure to send all hematologic labs before starting plasma exchange.
Guide to emoji hyperlinks 
 = Link to online calculator.
= Link to online calculator. = Link to Medscape monograph about a drug.
= Link to Medscape monograph about a drug. = Link to IBCC section about a drug.
= Link to IBCC section about a drug. = Link to IBCC section covering that topic.
= Link to IBCC section covering that topic. = Link to FOAMed site with related information.
= Link to FOAMed site with related information. = Link to supplemental media.
= Link to supplemental media.
References
- 09299856 Bobbio-Pallavicini E, Gugliotta L, Centurioni R, Porta C, Vianelli N, Billio A, Tacconi F, Ascari E. Antiplatelet agents in thrombotic thrombocytopenic purpura (TTP). Results of a randomized multicenter trial by the Italian Cooperative Group for TTP. Haematologica. 1997 Jul-Aug;82(4):429-35 [PubMed]
- 26418759 Mariotte E, Veyradier A. Thrombotic thrombocytopenic purpura: from diagnosis to therapy. Curr Opin Crit Care. 2015 Dec;21(6):593-601. doi: 10.1097/MCC.0000000000000255 [PubMed]
- 27766045 Shen YM. Clinical evaluation of thrombotic microangiopathy: identification of patients with suspected atypical hemolytic uremic syndrome. Thromb J. 2016 Oct 4;14(Suppl 1):19. doi: 10.1186/s12959-016-0114-0 [PubMed]
- 28416507 Joly BS, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura. Blood. 2017 May 25;129(21):2836-2846. doi: 10.1182/blood-2016-10-709857 [PubMed]
- 29296701 Page EE, Kremer Hovinga JA, Terrell DR, Vesely SK, George JN. Thrombotic thrombocytopenic purpura: diagnostic criteria, clinical features, and long-term outcomes from 1995 through 2015. Blood Adv. 2017 Apr 6;1(10):590-600. doi: 10.1182/bloodadvances.2017005124 [PubMed]
- 29348275 Elayoubi J, Donthireddy K, Nemakayala DR. Microangiopathies in pregnancy. BMJ Case Rep. 2018 Jan 17;2018:bcr2017221648. doi: 10.1136/bcr-2017-221648 [PubMed]
- 29582550 Fox LC, Cohney SJ, Kausman JY, et al. Consensus opinion on diagnosis and management of thrombotic microangiopathy in Australia and New Zealand. Intern Med J. 2018 Jun;48(6):624-636. doi: 10.1111/imj.13804 [PubMed]
- 30031798 Sridharan M, Go RS, Willrich MAV. Atypical hemolytic uremic syndrome: Review of clinical presentation, diagnosis and management. J Immunol Methods. 2018 Oct;461:15-22. doi: 10.1016/j.jim.2018.07.006 [PubMed]
- 30865166 Hanna RM, Barsoum M, Vandross A, Kurtz I, Burwick R. Atypical hemolytic uremic syndrome and complement blockade: established and emerging uses of complement inhibition. Curr Opin Nephrol Hypertens. 2019 May;28(3):278-287. doi: 10.1097/MNH.0000000000000499 [PubMed]
- 30145224 Tsai HM. Atypical Hemolytic Uremic Syndrome: Beyond Hemolysis and Uremia. Am J Med. 2019 Feb;132(2):161-167. doi: 10.1016/j.amjmed.2018.08.011 [PubMed]
- 30294946 Raina R, Krishnappa V, Blaha T, Kann T, Hein W, Burke L, Bagga A. Atypical Hemolytic-Uremic Syndrome: An Update on Pathophysiology, Diagnosis, and Treatment. Ther Apher Dial. 2019 Feb;23(1):4-21. doi: 10.1111/1744-9987.12763 [PubMed]
- 30622684 Wiernek SL, Jiang B, Gustafson GM, Dai X. Cardiac implications of thrombotic thrombocytopenic purpura. World J Cardiol. 2018 Dec 26;10(12):254-266. doi: 10.4330/wjc.v10.i12.254 [PubMed]
- 31588978 Azoulay E, Bauer PR, Mariotte E, et al. Expert statement on the ICU management of patients with thrombotic thrombocytopenic purpura. Intensive Care Med. 2019 Nov;45(11):1518-1539. doi: 10.1007/s00134-019-05736-5. Epub 2019 Oct 7. Erratum in: Intensive Care Med. 2020 Mar;46(3):570-571 [PubMed]
- 31935318 Lee H, Kang E, Kang HG, et al. Consensus regarding diagnosis and management of atypical hemolytic uremic syndrome. Korean J Intern Med. 2020 Jan;35(1):25-40. doi: 10.3904/kjim.2019.388 [PubMed]
- 32172817 Manrique-Caballero CL, Peerapornratana S, Formeck C, Del Rio-Pertuz G, Gomez Danies H, Kellum JA. Typical and Atypical Hemolytic Uremic Syndrome in the Critically Ill. Crit Care Clin. 2020 Apr;36(2):333-356. doi: 10.1016/j.ccc.2019.11.004 [PubMed]
- 32259874 Blennerhassett R, Curnow J, Pasalic L. Immune-Mediated Thrombotic Thrombocytopenic Purpura: A Narrative Review of Diagnosis and Treatment in Adults. Semin Thromb Hemost. 2020 Apr;46(3):289-301. doi: 10.1055/s-0040-1708541 [PubMed]
- 32539032 Htet Z, Karim M. Thrombotic microangiopathy with renal injury: an approach for the general physician. J R Coll Physicians Edinb. 2020 Mar;50(1):25-31. doi: 10.4997/JRCPE.2020.107 [PubMed]
- 32757237 Paydary K, Banwell E, Tong J, Chen Y, Cuker A. Diagnostic accuracy of the PLASMIC score in patients with suspected thrombotic thrombocytopenic purpura: A systematic review and meta-analysis. Transfusion. 2020 Sep;60(9):2047-2057. doi: 10.1111/trf.15954 [PubMed]
- 32808006 Fakhouri F, Scully M, Provôt F, et al. Management of thrombotic microangiopathy in pregnancy and postpartum: report from an international working group. Blood. 2020 Nov 5;136(19):2103-2117. doi: 10.1182/blood.2020005221 [PubMed]
- 32914582 Zheng XL, Vesely SK, Cataland SR, Coppo P, Geldziler B, Iorio A, Matsumoto M, Mustafa RA, Pai M, Rock G, Russell L, Tarawneh R, Valdes J, Peyvandi F. ISTH guidelines for the diagnosis of thrombotic thrombocytopenic purpura. J Thromb Haemost. 2020 Oct;18(10):2486-2495. doi: 10.1111/jth.15006 [PubMed]
- 32914526 Zheng XL, Vesely SK, Cataland SR, Coppo P, Geldziler B, Iorio A, Matsumoto M, Mustafa RA, Pai M, Rock G, Russell L, Tarawneh R, Valdes J, Peyvandi F. ISTH guidelines for treatment of thrombotic thrombocytopenic purpura. J Thromb Haemost. 2020 Oct;18(10):2496-2502. doi: 10.1111/jth.15010 [PubMed]
- 32950988 Avila Bernabeu AI, Cavero Escribano T, Cao Vilarino M. Atypical Hemolytic Uremic Syndrome: New Challenges in the Complement Blockage Era. Nephron. 2020;144(11):537-549. doi: 10.1159/000508920 [PubMed]
- 33179792 Liu A, Dhaliwal N, Upreti H, Kasmani J, Dane K, Moliterno A, Braunstein E, Brodsky R, Chaturvedi S. Reduced sensitivity of PLASMIC and French scores for the diagnosis of thrombotic thrombocytopenic purpura in older individuals. Transfusion. 2021 Jan;61(1):266-273. doi: 10.1111/trf.16188 [PubMed]
- 33540569 Sukumar S, Lämmle B, Cataland SR. Thrombotic Thrombocytopenic Purpura: Pathophysiology, Diagnosis, and Management. J Clin Med. 2021 Feb 2;10(3):536. doi: 10.3390/jcm10030536 [PubMed]
- 33841853 Blasco M, Guillén E, Quintana LF, Garcia-Herrera A, Piñeiro G, Poch E, Carreras E, Campistol JM, Diaz-Ricart M, Palomo M. Thrombotic microangiopathies assessment: mind the complement. Clin Kidney J. 2020 Nov 6;14(4):1055-1066. doi: 10.1093/ckj/sfaa195 [PubMed]
